博文
代谢学人—Nature:线粒体融合的守护神
||
代谢学人
Nature:线粒体融合的守护神
撰文 | 陈芳芳 胡敏 马莹 张俊 武霞 郭明伟
编辑 | 孟美瑶
校对 | 张俊

背景介绍
细胞在面对许多病原体和外在压力时,可以通过降解错误折叠的蛋白质,通过溶酶体清除全部或部分线粒体,以及通过调节转录和翻译层面上线粒体蛋白质的水平来保护线粒体。线粒体应激反应受损与神经变性、心力衰竭和代谢综合征有关。然而,这些应激反应途径在生理条件下的作用仍然难以捉摸。线粒体功能障碍会诱导由Parkin-PINK1和OMA1介导的两种压力传感系统。Parkin是一种泛素E3连接酶,在膜电位丧失后被PINK1介导的磷酸化激活并被募集到线粒体外膜。Parkin能够泛素化参与线粒体融合、降解、线粒体来源的囊泡和生物合成的线粒体蛋白。Parkin和PINK1的缺陷会导致偶发性和家族性帕金森病。OMA1是线粒体内膜上的一种金属蛋白酶,与Parkin-PINK1相似,OMA1被降低的线粒体膜电位激活。激活的OMA1切割线粒体融合GTP酶(GTPase)——视神经萎缩蛋白1(opticatrophy1,OPA1),抑制线粒体融合。活化的OMA1还能裂解并释放线粒体蛋白DAP3结合细胞死亡增强因子1(DELE1)到细胞质中,刺激整合应激反应(ISR)。而Parkin、PINK1或OMA1基因敲除(KO)小鼠在正常生理状态下,线粒体功能和动物生理均无明显缺陷(图1a-d,e(i,ii))。这就表明Parkin-PINK1和OMA1可能主要在响应外部压力时发挥作用,因此在非应激状态下是非必需的。或者,即使在正常生理条件下,这两个压力传感系统也可能具有关键的功能,但一个防御系统的损失可以由另一个系统来补偿。

敲黑板啦!
1.Parkin和OMA1单独敲除对小鼠无影响;
2.Parkin-PINK1和OMA1对线粒体融合进行双重调控;
3.Parkin和OMA1并不影响代谢;
4.大脑是受Parkin和OMA1双重敲除影响最大的组织;
5.Parkin和OMA1对运动神经元具有保护作用。
研究结果
1 .Parkin - PINK1和OMA1确保小鼠健康
为了验证上述两个假设,研究人员构建了Parkin和OMA1的全身纯合双基因敲除小鼠。值得注意的是,研究人员发现Parkin-/-Oma1-/-小鼠非常小,过早死亡,平均存活约70天(图1a-c,e(iii))。在旷场实验中,Parkin-/-Oma1-/-小鼠的整体运动能力也显著下降(图1d)。肉眼观察发现,Parkin-/-Oma1-/-小鼠出现抽搐现象,而野生型(WT)和单基因敲除(single-KO)小鼠均未出现此表型。为了确定PINK1的缺失是否具有类似于Parkin的作用,研究人员构建了Pink1-/-Oma1-/-小鼠。Pink1-/-Oma1-/-小鼠表型与Parkin-/-Oma1-/-小鼠相似,表现为体形矮小,早期死亡,旷场实验中活动减少(图1a-d,e(iii))。这些数据表明,Parkin–PINK1和OMA1协同保护小鼠的发育、生理和生存。
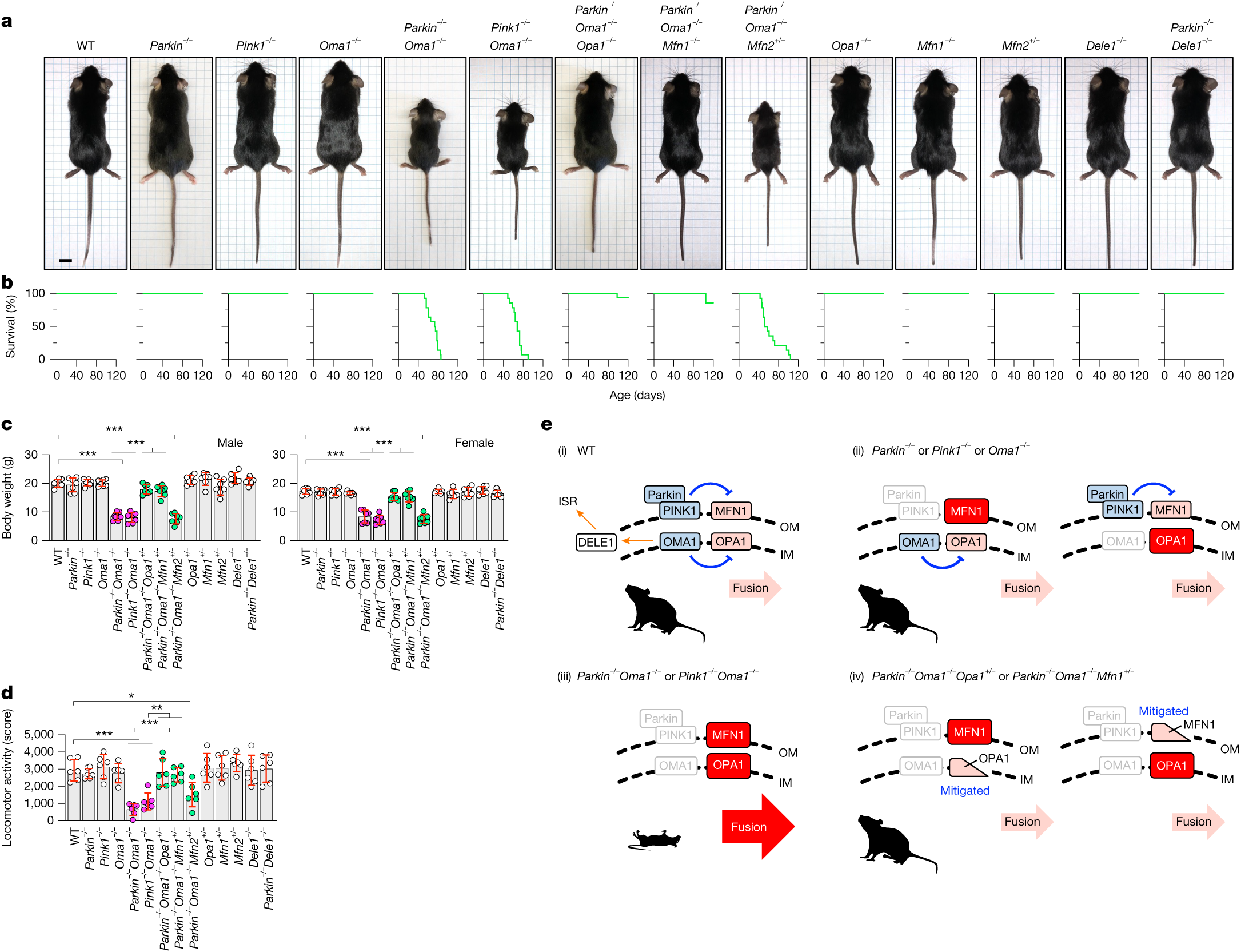
图1 | Parkin - PINK1和OMA1通过抑制线粒体融合来保证小鼠的健康
2 .Parkin和OMA1平衡线粒体动力学
在线粒体融合过程中,金属蛋白酶OMA1水解OPA1失活,使其失活。为了确定OPA1是否对Parkin-/-Oma1-/-表型至关重要,研究人员在Parkin-/-Oma1-/-小鼠中引入了OPA1的杂合KO(Parkin-/-Oma1-/-Opa1+/-),因为纯合的OPA1 KO是胚胎致死的。值得注意的是,杂合子OPA1缺失显著改善了Parkin-/-Oma1-/-小鼠的体型、存活率和运动能力(图1a-d,e(iv))。OPA1介导线粒体融合和内膜嵴的形成。为了分离OPA1的这两种功能,研究人员在Parkin-/-Oma1-/-小鼠中引入了MFN1的杂合子KO,MFN1控制线粒体融合,但不控制嵴结构。研究人员发现Parkin-/-Oma1-/-Mfn1+/-小鼠与Parkin-/-Oma1-/-Opa1+/-小鼠表型相似(图1a-d,e(iv))。这些发现表明,在体内没有外界线粒体应激的情况下,Parkin和OMA1调控OPA1-MFN1融合机制。与MFN1不同的是, MFN1同源基因MFN2的杂合缺失并不能改善Parkin-/-Oma1-/-小鼠的表型(图1a-d)。这些数据与之前的研究一致,表明OPA1与MFN1有效地介导线粒体融合。
为了验证参与ISR的另一个OMA1底物DELE1的作用,研究人员利用CRISPR技术构建了全身敲除的Dele1-/-小鼠,并与Parkin-/-小鼠杂交(扩展数据图1)。如果Parkin-/-Oma1-/-小鼠的综合表型是由Parkin和OMA1共同缺失引起的ISR介导的,那么Parkin-/-Dele1-/-小鼠应该与Parkin-/-Oma1-/-小鼠表型一致。然而,Dele1-/-和Parkin-/-Dele1-/-小鼠在体形、存活和运动能力方面是正常的(图1a-d)。这些数据表明Parkin-/-Oma1-/-小鼠的缺陷与线粒体融合的增加有关,而不是与ISR的抑制有关。
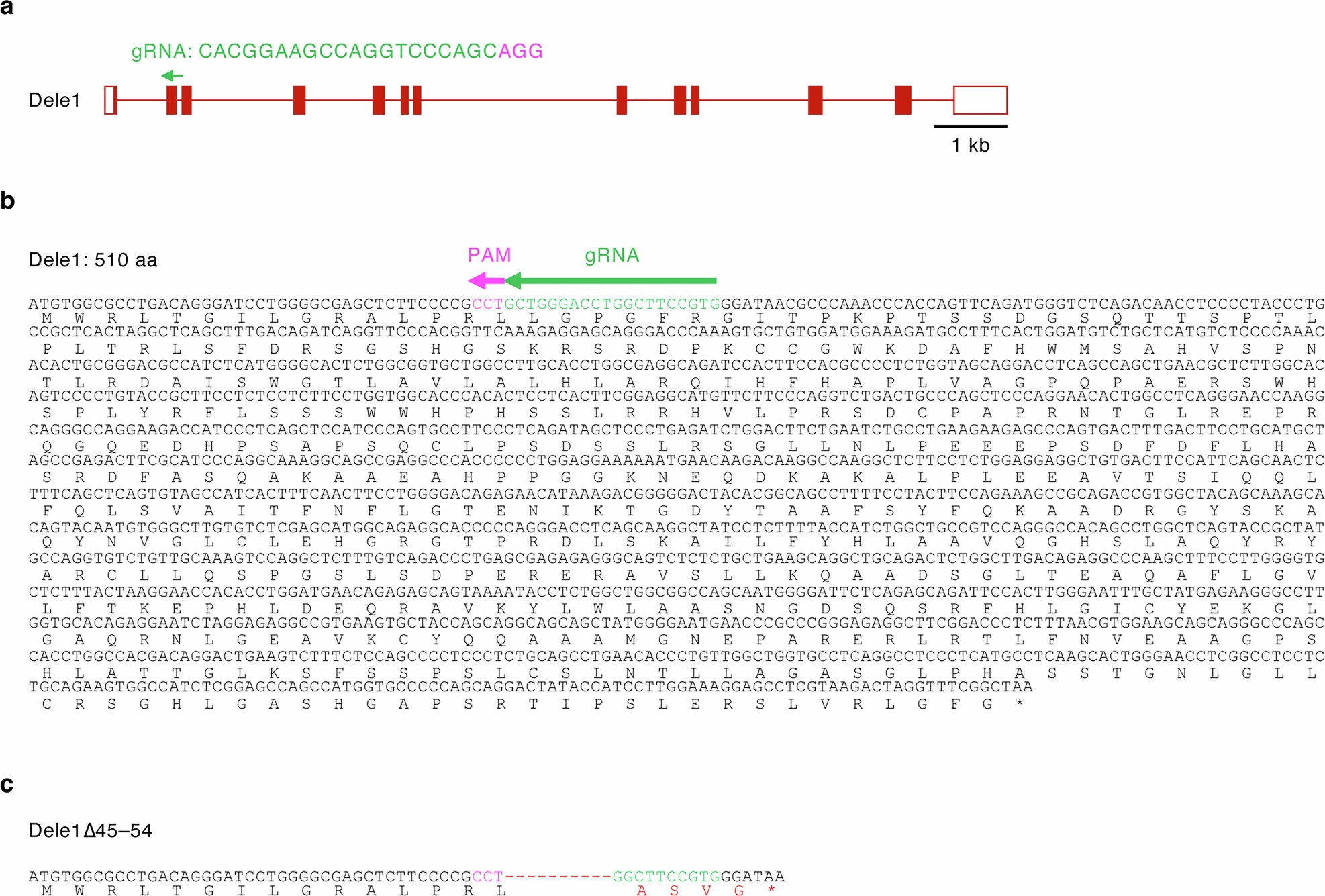
扩展数据图1 | Dele1-/-小鼠的产生
3 . Parkin和OMA1防止过度融合
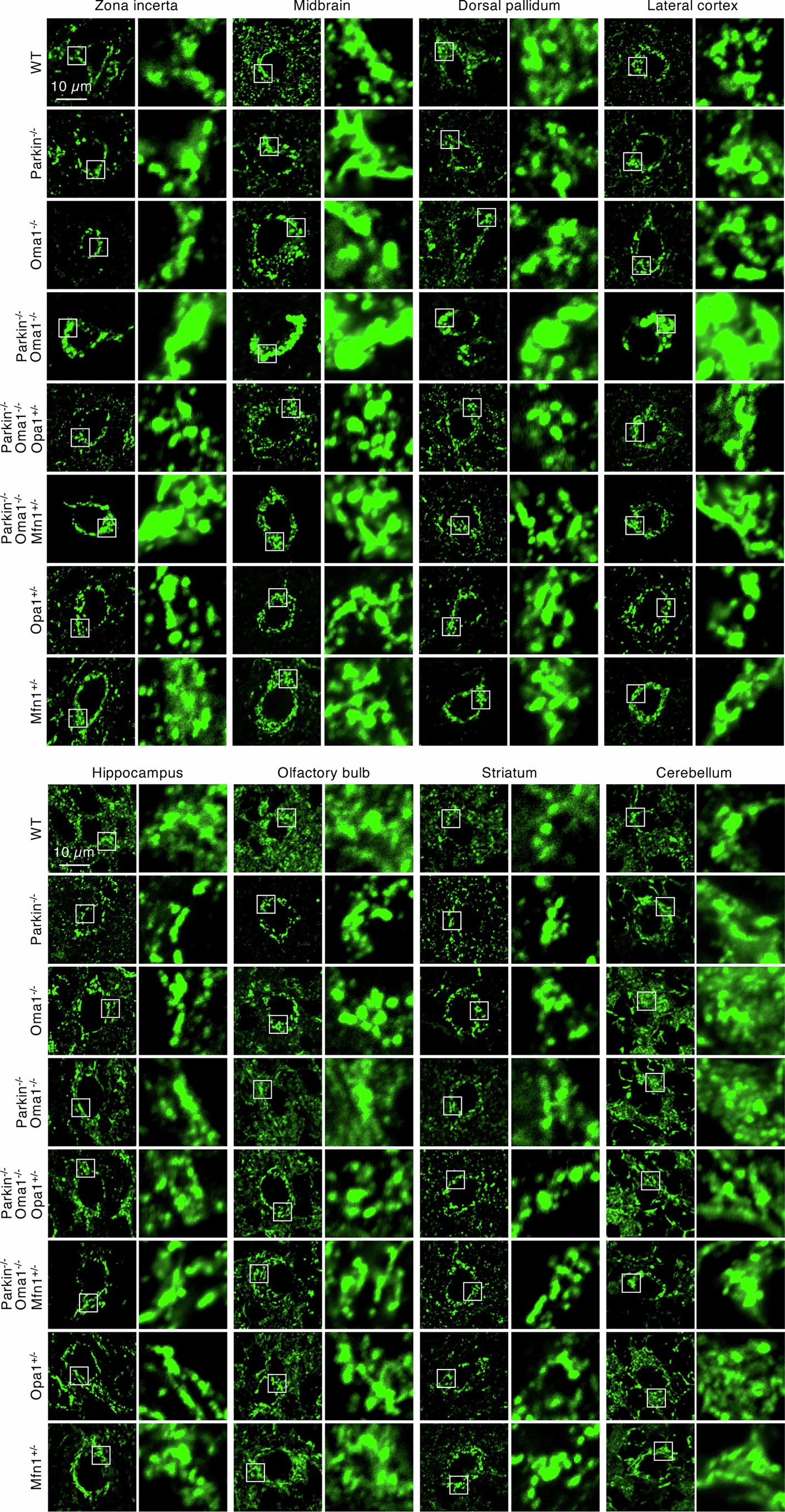
为了确定Parkin和OMA1的缺失是否影响线粒体形态,研究人员开发了系统的线粒体形态分析方法,利用线粒体蛋白丙酮酸脱氢酶(pyruvatedehydrogenase,PDH)的抗体,使用激光共聚焦免疫荧光显微镜评估了10个大脑亚区和8个主要器官(心、肾、棕色脂肪组织、骨骼肌、肺、肝、肠、脾)的线粒体形态(图2a-c和扩展数据图2-4)。
系统的线粒体形态分析表明,Parkin或OMA1的单KO不影响线粒体形态(图2a-c和扩展数据图2-4)。相比之下,Parkin-/-Oma1-/-小鼠在多个器官中观察到线粒体增大(巨线粒体),在大脑和心脏中观察到的效应最强(图2a-c和扩展数据图2-4)。在大脑内,脑桥/延髓显示出最高频率的巨线粒体形成,其次是未定带,中脑和背侧苍白球(图2b,c和扩展数据图4)。当研究人员单独分析脑桥和延髓的巨线粒体时,这两个区域中巨线粒体的数量是相似的(扩展数据图5a)。在脑桥和延髓中,大多数含有巨线粒体的细胞表现出神经元标记物NeuN阳性,而对小胶质细胞(Iba1)、星形胶质细胞(GFAP)或血管细胞(PECAM1)的标记物呈阴性,表明脑桥/延髓神经元对的线粒体形态调控高度依赖Parkin和OMA1(扩展数据图5b-e)。此外,透射电镜显示,在脑桥/延髓和心脏中,增大的线粒体含有较少的内膜嵴(图2d)。值得注意的是,在Parkin-/-Oma1-/-小鼠中,OPA1(Parkin-/-Oma1-/-Opa1+/− )或MFN1的部分减少( Parkin-/-Oma1-/-Mfn1+/−)改善了Parkin-/-Oma1-/-小鼠线粒体形态增大和嵴结构紊乱(图2a-d和扩展数据图4)。因此,Parkin-/-Oma1-/-小鼠的线粒体动力学平衡向融合方向倾斜。这些数据表明Parkin和OMA1在生理条件下阻止了过度的线粒体融合。
免疫印迹分析显示,在Parkin-/-小鼠的脑桥/延髓、小脑和肝脏中,MFN1的水平上调幅度不大,但具有显著性,而MFN2的水平没有显著变化(扩展数据图6a-d)。这种MFN1特异性的增加与在Parkin-/-Oma1-/-小鼠中引入MFN1杂合KO改善了Parkin-/-Oma1-/-小鼠表型结果一致(图1a-d)。研究人员还发现,与WT小鼠相比,Oma1-/-小鼠的脑桥/延髓、小脑和肝脏中对OPA1的处理减少了(小编注:这里说的是OPA1被OMA1裂解的产物减少了,OPA1有两种较长的亚型(L1/L2),而金属蛋白酶OMA1会裂解OPA1两种较长的亚型, L1会被OMA1裂解成为S3亚型,L2会被裂解成为S5亚型,扩展数据图6f中对OPA1的亚型进行定量,L2亚型在Oma1敲除之后上调,而S3和S5亚型均在Oma1敲除之后上调,就说明OMA1对OPA1的裂解减少了,也就是对OPA1的处理减少。)(扩展数据图6e,f)。这些数据表明Parkin和OMA1在这些组织中调节MFN1和OPA1;然而,不同组织之间的表型结果,特别是线粒体增大存在差异。与OPA1相反,在Oma1-/-小鼠的脑桥/延髓、小脑或肝脏中,OMA1的另一个底物Pgam5的蛋白水解过程不受影响,这与之前的研究一致,即在没有另一个内膜蛋白酶PARL的情况下,Oma1可在线粒体应激下切割Pgam5(扩展数据图6e,f)。
扩展阅读:OMA1对底物的调控,包括DELE,OPA1,PGAM5
OMA1可以通过调控内膜蛋白OPA1蛋白的水解,起到控制细胞线粒体分裂和融合的动态平衡。在正常状态下,OPA1被组成型蛋白酶(如YME1L)部分切割,维持L-OPA1/S-OPA1平衡。发生线粒体应激时,OMA1被激活,会快速切割L-OPA1,从而导致S-OPA1积累,抑制线粒体融合,促进碎片化以适应应激或启动凋亡。
正常情况下,DELE1被不断导入线粒体并被蛋白酶加工,在线粒体深处,DELE1会被线粒体基质驻留蛋白酶LONP1迅速降解。当这个导入过程因线粒体应激而失败时,新的DELE1分子在进入线粒体的过程中被阻止,DELE1则可能被OMA1切断形成短片段S-DELE1,或者在细胞器外保持未分裂状态,从而激活HRI介导的综合应激反应(ISR)保护细胞免受应激损伤。
磷酸甘油酸变位酶5(PGAM5)是一种线粒体Ser/Thr蛋白磷酸酶,在其N末端跨膜域中被切割以响应线粒体膜电位损失,进而促进细胞坏死性凋亡。这种线粒体膜电位损失依赖的PGAM5裂解是由菱形蛋白酶(PARL)介导的。线粒体膜电位丧失时,PARL是PGAM5的主要剪切酶,但OMA1在PARL缺失时可代偿性切割PGAM5。
参考文献:
[1] Pedro M, et al. Adipocyte. 2013.
[2] Evelyn F, et al.Nature Communications. 2022.
[3] Yusuke S, et al. Molecular Cell. 2023.
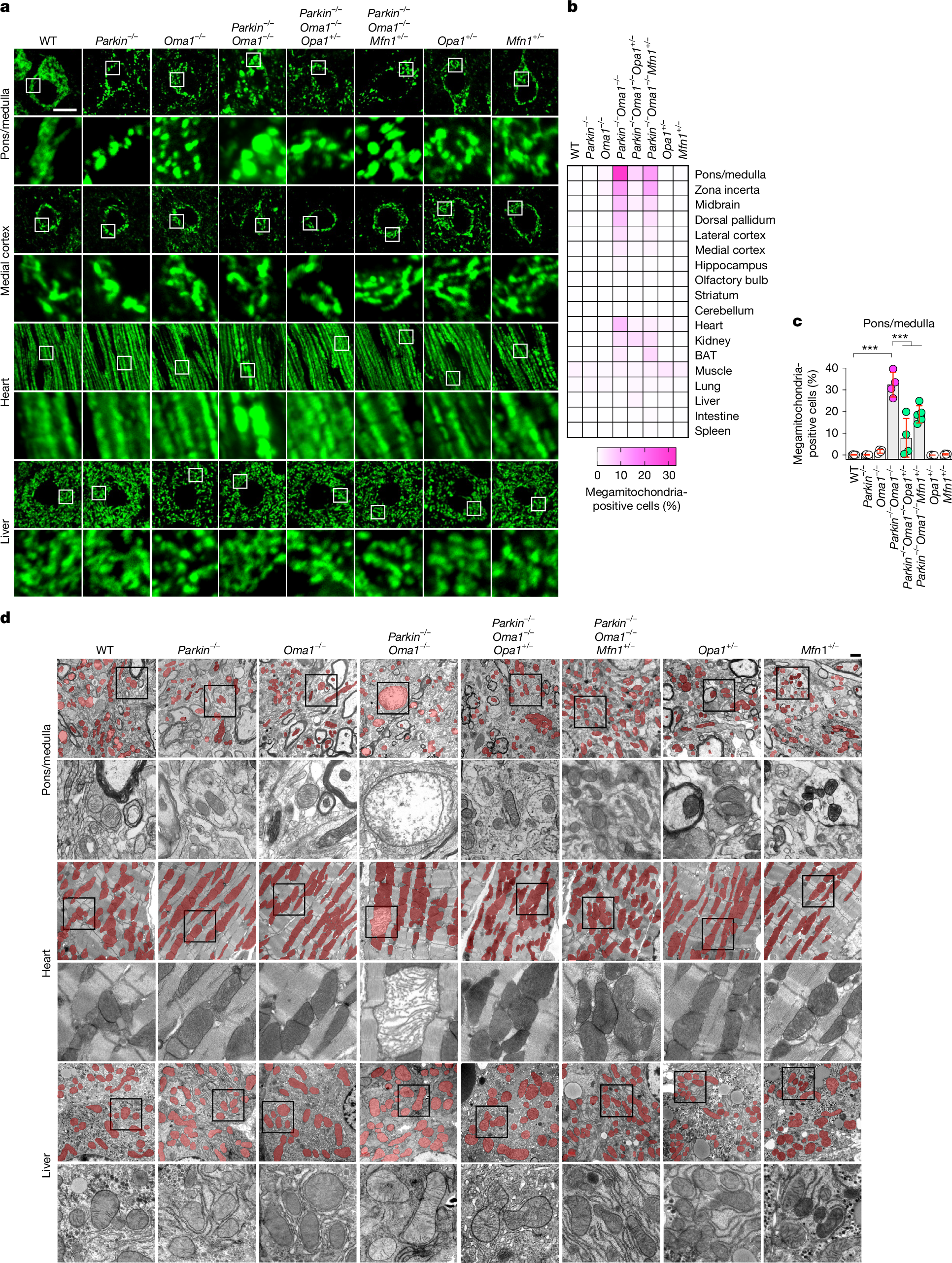
图2 | Parkin和OMA1阻止过度的线粒体融合
扩展数据图2 | 线粒体在未定带、中脑、背侧苍白球、外侧皮质、海马、嗅球、纹状体和小脑的代表性图像
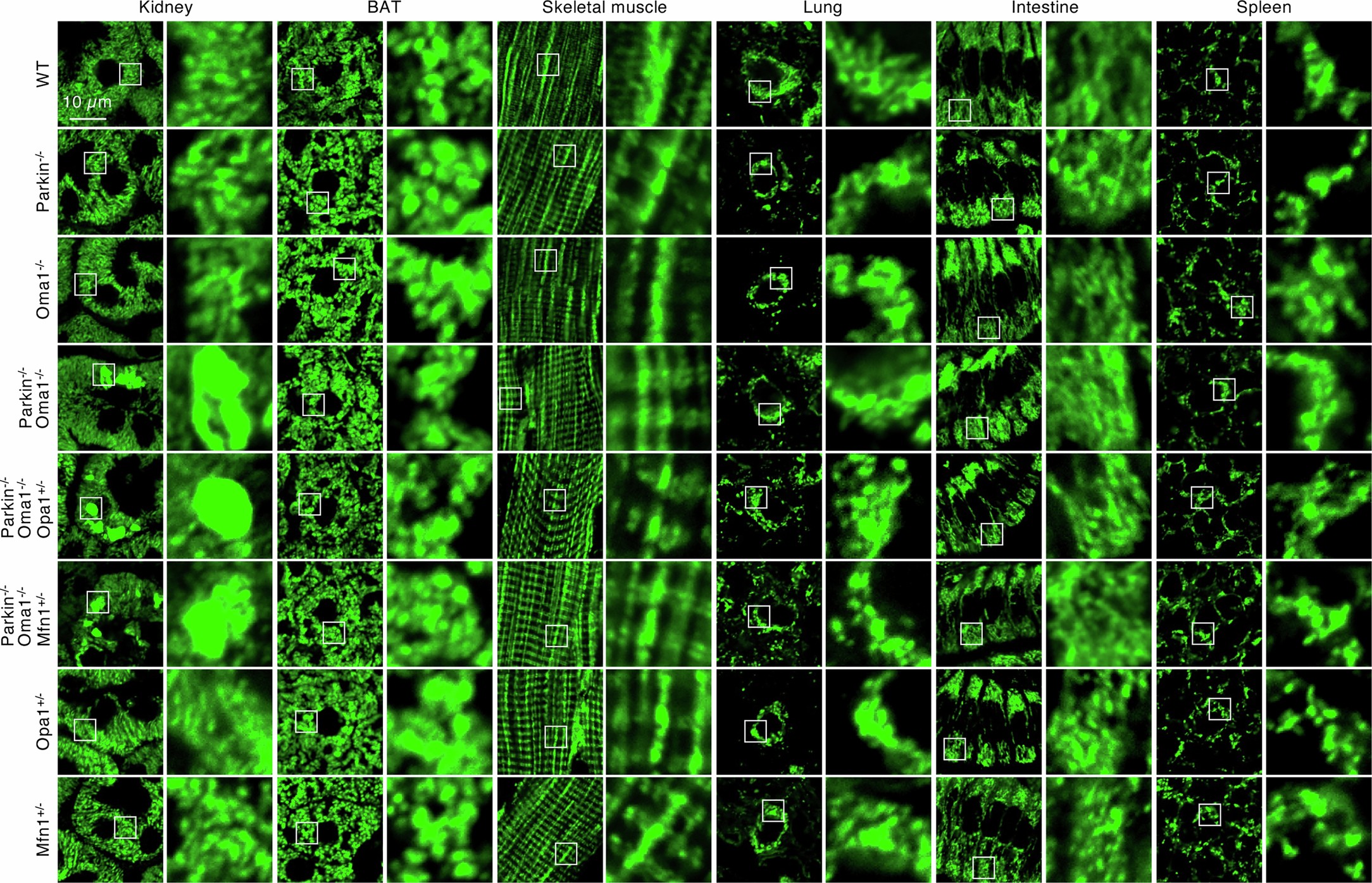
扩展数据图3 | 线粒体在肾脏、棕色脂肪组织(BAT)、骨骼肌、肺、肠和脾脏中的代表性图像
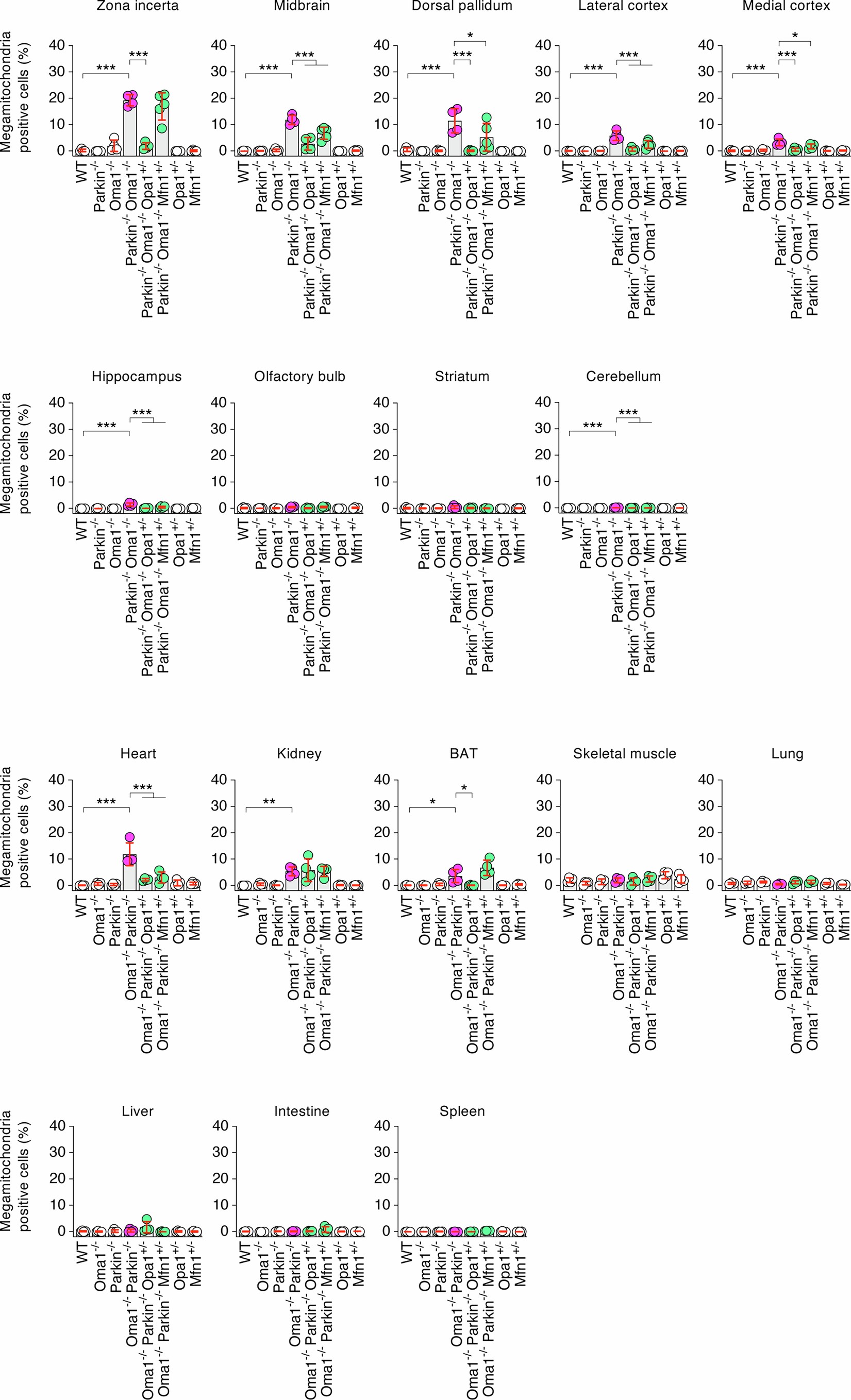
扩展数据图4 | 对含有增大线粒体的细胞进行定量
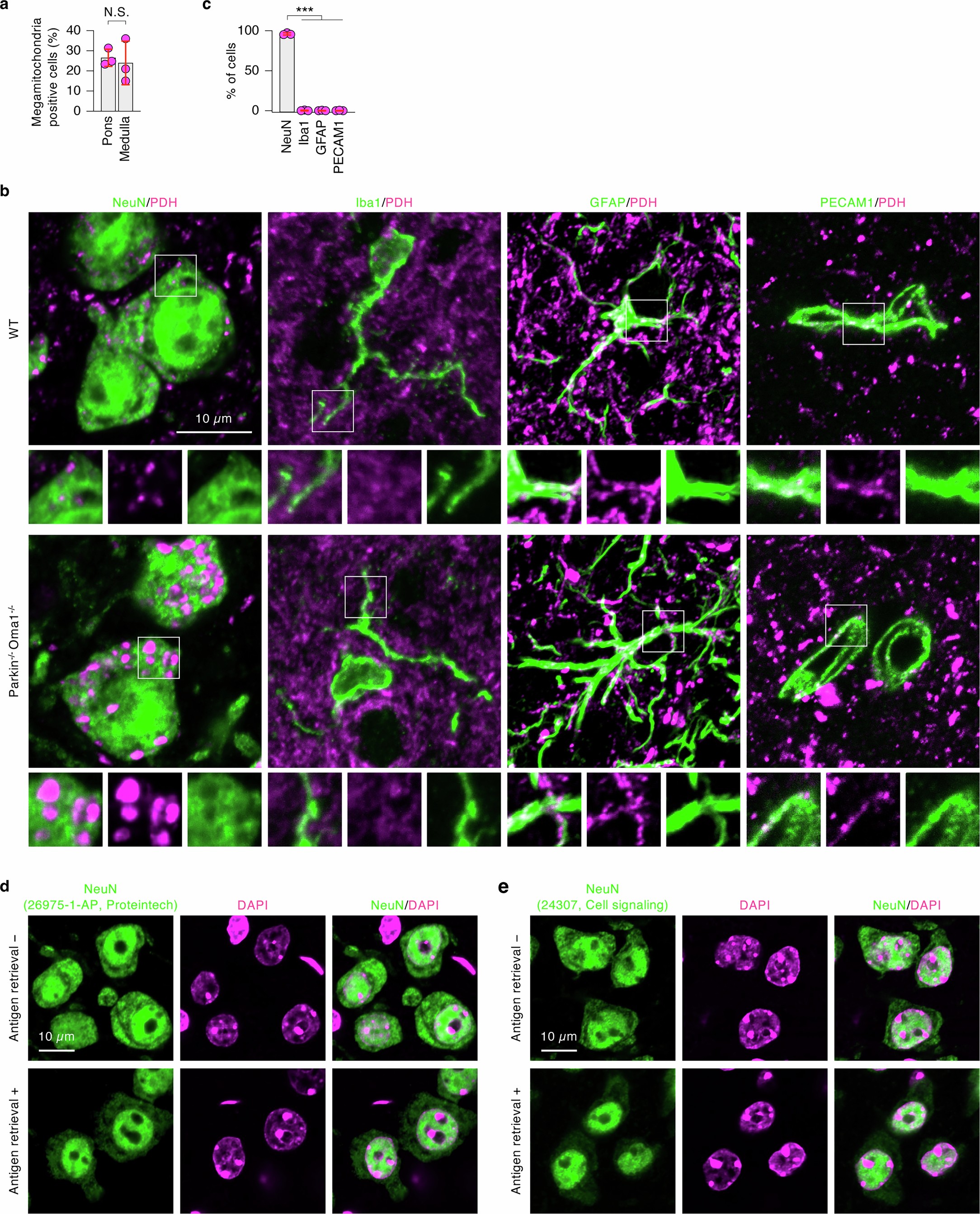
扩展数据图5 | 巨线粒体在Parkin-/-Oma1-/-小鼠的神经元中形成
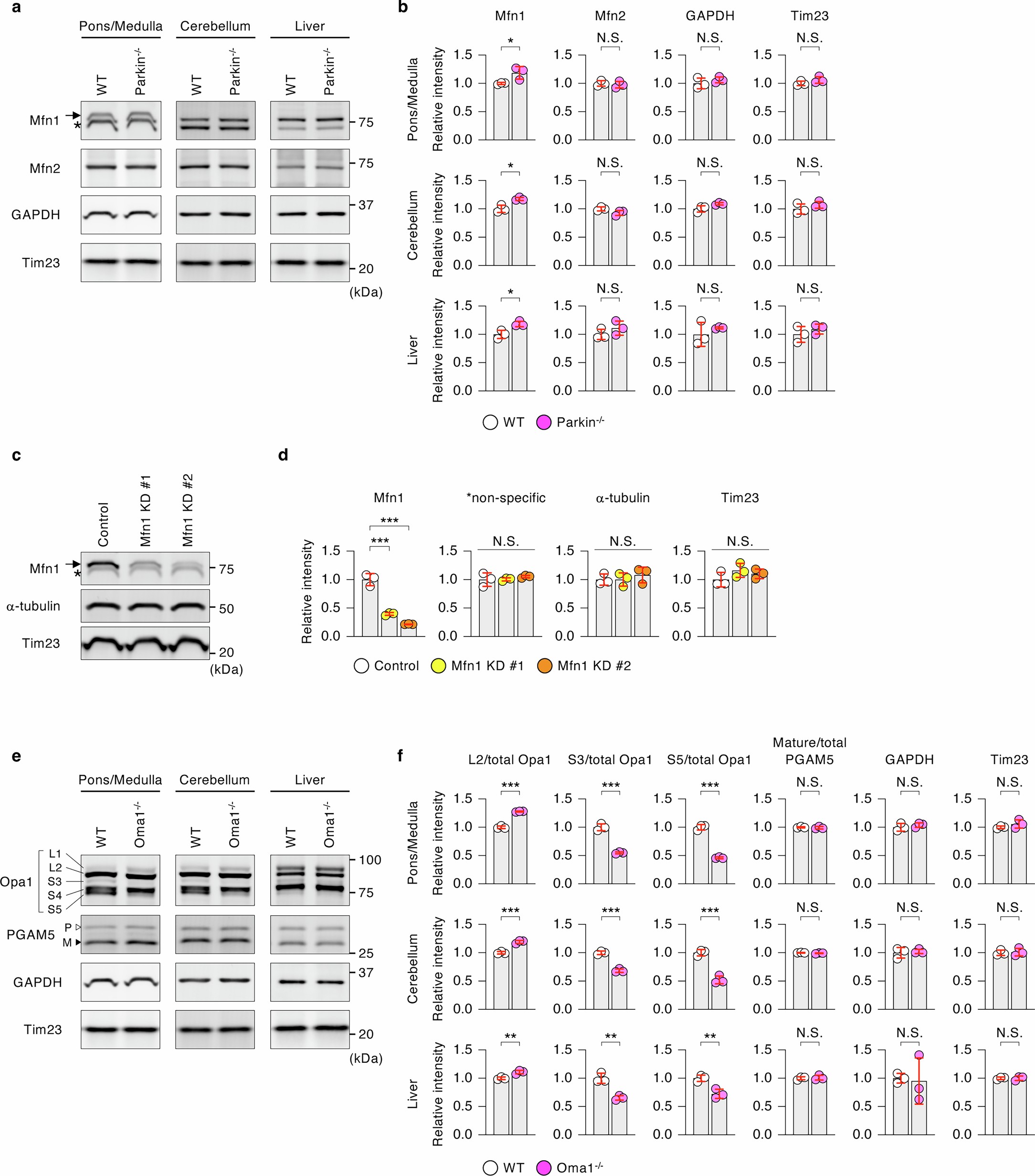
扩展数据图6 | Western印迹分析
4 .Parkin和OMA1不会影响代谢
为了研究Parkin和OMA1的联合缺失如何影响细胞代谢,研究人员首先使用液相色谱-质谱联用技术对WT和Parkin-/-Oma1-/-小鼠的脑桥/延髓进行了非靶向代谢组学研究。研究人员鉴定了188种代谢物,研究人员发现,与WT小鼠脑桥/延髓相比,这些代谢物在Parkin-/-Oma1-/-小鼠脑桥/延髓中和没有显著变化(扩展数据图7a)。这些代谢物包括ATP、ADP、AMP、三羧酸(TCA)循环代谢物以及许多参与合成代谢过程的物质(扩展数据图7b)。其次,研究人员分别检测了9种TCA循环酶的活性,这些酶在WT和Parkin-/-Oma1-/-小鼠脑桥/髓质中均显示出相似的活性(扩展数据图7c)。第三,为了分析线粒体呼吸,研究人员测量了WT和Parkin-/-Oma1-/-小鼠大脑的耗氧率(OCR);Parkin和OMA1的丢失不影响OCR(扩展数据图7d)(小编注:在第一个阶段加入琥珀酸(succinate)是为了检测线粒体复合物Ⅱ的功能,琥珀酸是线粒体复合物Ⅱ的特异性底物,可以直接反应其活性,如果复合物Ⅱ功能受损,则加入琥珀酸之后OCR不会显著增加;第二节阶段加入抗霉素A(antimycin A)和鱼藤酮(rotenone)分别是线粒体复合物Ⅲ和复合物Ⅰ的抑制剂,这两种物质能够关闭线粒体呼吸,从而计算线粒体之外活动所驱动的非线粒体呼吸耗氧;第三个阶段加入的四甲基对苯二胺(TMPD)和抗坏血酸(Asc)是为了评估线粒体复合物Ⅳ(细胞色素c氧化酶,COX)的功能,Asc是一种还原剂,能够将细胞色素c还原为还原态,而还原态的细胞色素c是复合物Ⅳ的直接电子供体,TMPD能够增强电子从抗坏血酸到细胞色素c的传递效率,加入TMPD和Asc之后引起OCR增加,表明复合物Ⅳ功能正常;在经典的OCR实验中,叠氮化钠(Azide)通常作为最后一步抑制剂,用于抑制复合物IV,确认其对OCR的贡献,其次也是为了验证整个电子传递链是否已被完全阻断,加入Azide之后OCR下降,表明复合物Ⅳ是之前氧气消耗的主要驱动力。)。这些数据表明,Parkin-/-Oma1-/-小鼠的线粒体代谢没有明显改变。
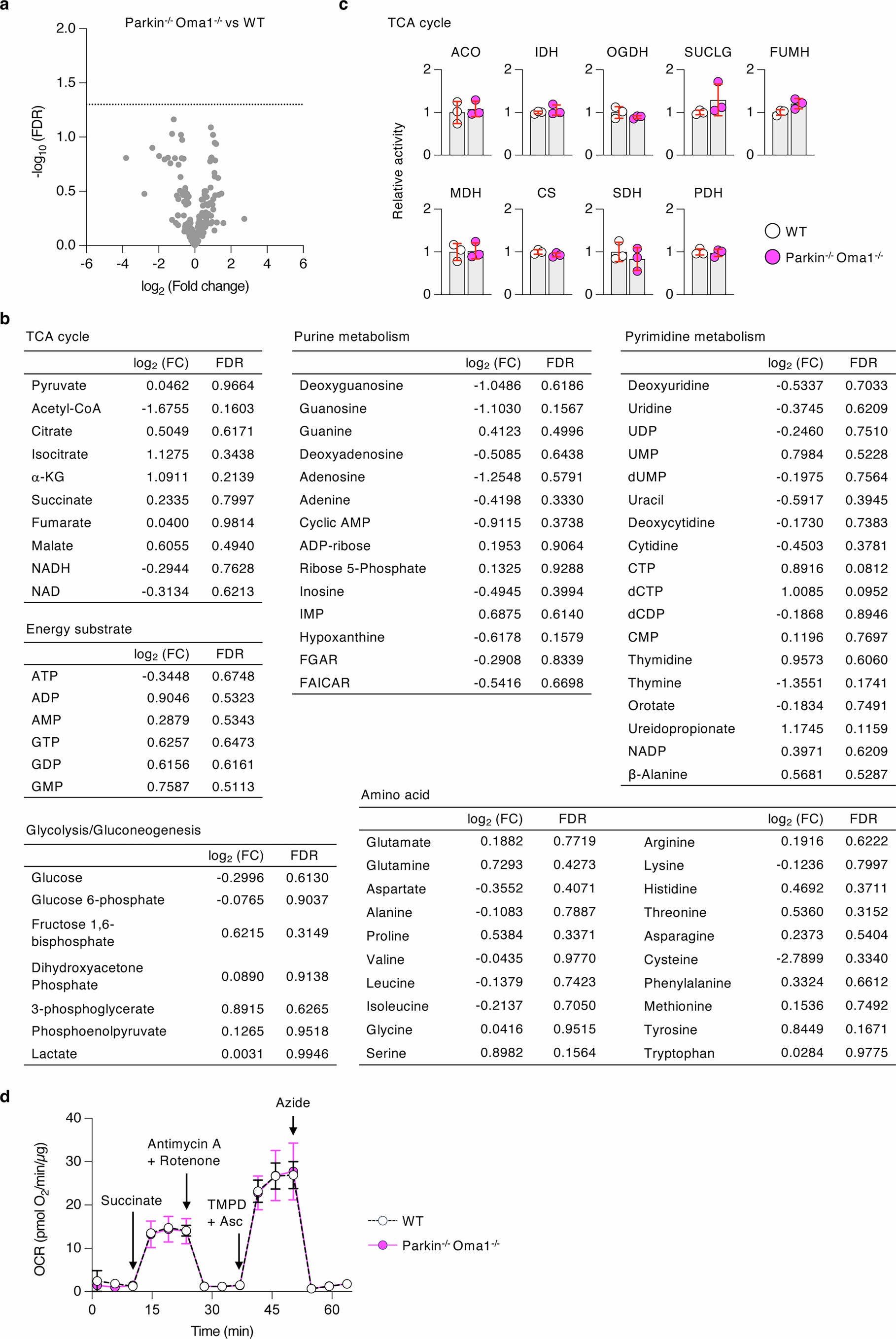
扩展数据图7 | 代谢物组景观的比较
5 .Parkin和OMA1保护线粒体基因组
为了揭示Parkin和OMA1的缺失对代谢以外的影响,研究人员对WT、Parkin-/-Oma1-/-、Parkin-/-Oma1-/-Opa1+/小鼠-和Parkin-/-Oma1-/-Mfn1+/-小鼠脑桥/延髓进行了转录组测序(RNA-seq)。基因集富集分析(GSEA)显示,与WT小鼠相比,Parkin-/-Oma1-/小鼠-脑桥/延髓中参与免疫反应的基因上调(图3a,b,扩展数据图8a)。在前10个条目中,研究人员用GO分析的生物学过程标记了9条通路,用MSigDB Hallmark pathway数据库标注了与免疫反应有关的7条通路(小编注:研究人员在这里用两种方式分析了GSEA,一种方式是GO分析中的生物过程,另一种方式是MSigDB Hallmark pathway,MSigDB是一个汇集了经过良好注释的基因集合的数据库,被广泛用于分析基因富集通路,其中Hallmark pathway总结了特定的明确定义的生物状态或过程。)。根据这些发现,研究人员收集了与16条富集通路相关的所有差异表达基因(图3a),并对差异表达基因进行下一步分析(图3b)。在Parkin-/-Oma1-/-Opa1+/-和Parkin-/-Oma1-/-Mfn1+/-小鼠的脑桥/延髓中这些基因的表达模式恢复到接近WT小鼠水平(图3b),表明在Parkin-/-Oma1-/-小鼠脑桥/延髓中观察到的基因表达升高是由于过度融合而不是Parkin缺失引起的线粒体自噬缺陷导致的。脑桥/延髓的定量PCR(qPCR)分析显示干扰素刺激基因Ifit3、Usp18、Oasl2、Ddx60、Bst2和Sting1上调说明了Parkin-/-Oma1-/-小鼠先天性免疫反应的激活(扩展数据图9)。
由于线粒体DNA释放到细胞质中时,可以通过干扰素基因的蛋白刺激因子(STING)诱导先天免疫反应,研究人员使用激光共聚焦免疫荧光显微镜和图像定量检测了WT和Parkin-/-Oma1-/-小鼠脑桥/延髓中的mtDNA。研究人员发现,在WT小鼠脑桥/延髓的线粒体外的区域只有不到1%的DNA信号(图3c,e)。同样,与WT小鼠相比,在Parkin-/-Oma1-/-小鼠桥脑/延髓中具有正常线粒体形态的细胞显示出与WT小鼠相似的mtDNA大小和线粒体以外的DNA水平(图3c-e;正常线粒体)。相反,与WT小鼠相比,在Parkin-/-Oma1-/-小鼠的脑桥/延髓中线粒体增大的细胞中,线粒体外的区域的mtDNA大小和水平显著增加,上升至6-7%(图3c-e;巨形线粒体)。在这些增大的线粒体中,研究人员观察到DNA信号的强度增加,可能是由于mtDNA核苷酸的聚集造成的(图3c);这些线粒体外的信号大部分位于线粒体附近(图3c)。这些数据表明,在Parkin-/-Oma1-/-小鼠的脑桥/延髓中,mtDNA是由增大的线粒体释放的,而不是从正常形状的线粒体中释放出来的。
研究人员通过生物组分分离实验与qPCR的方法证实了Parkin-/-Oma1-/-小鼠脑桥/延髓中mtDNA释放到胞浆中(图3f)(小编注:D-loop区和16S rRNA是mtDNA中两个关键的区域。D-loop区域是mtDNA中的一段非编码区,位于线粒体基因组的调控区域,其包含三个主要的功能原件:H链启动子(Heavy Strand Promoter,HSP),负责重链的起始转录;L链启动子(Light Strand Promoter,LSP),负责轻链的转录起始;复制起点(OriH和OriL)分别控制重链和轻链的复制起始。16S rRNA是mtDNA中编码核糖体RNA的一个重要基因,16S rRNA是线粒体核糖体大亚基的核心组成部分,负责与蛋白质结合形成功能性核糖体。这里的16S和细菌中的16S相同,都是组成核糖体大亚基的核心部分)。通过免疫印迹证实了胞浆部分的均一性(图3g)。线粒体内的mtDNA水平在WT和Parkin-/-Oma1-/-小鼠桥脑/延髓之间相似(扩展数据图8b,c)。因此,只有相对较小部分的mtDNA被释放到细胞质中,这并不显著影响线粒体中mtDNA的整体含量。总之,这些数据表明释放的mtDNA激活了Parkin-/-Oma1-/-小鼠桥脑/延髓中的STING通路。为了了解STING在Parkin-/-Oma1-/-小鼠表型中的作用,研究人员利用STING缺陷的Stinggt/gt小鼠构建了三纯合突变小鼠Parkin-/-Oma1-/-Stinggt/gt。STING的额外缺失显著改善了Parkin-/-Oma1-/-小鼠的体型和死亡率(图3h-j),强调了STING在这些表型中的关键作用。
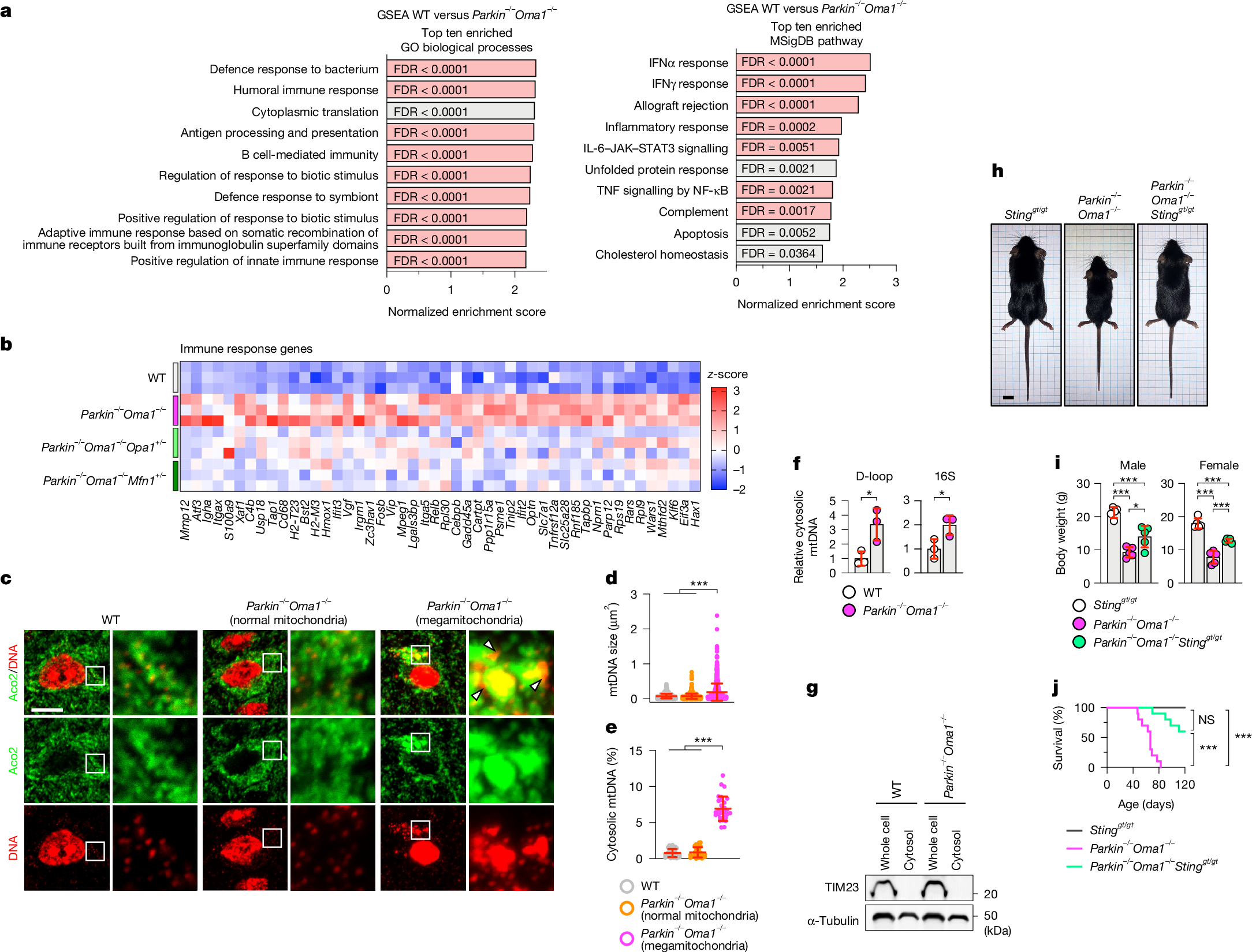
图3 | Parkin和OMA1保护线粒体基因组
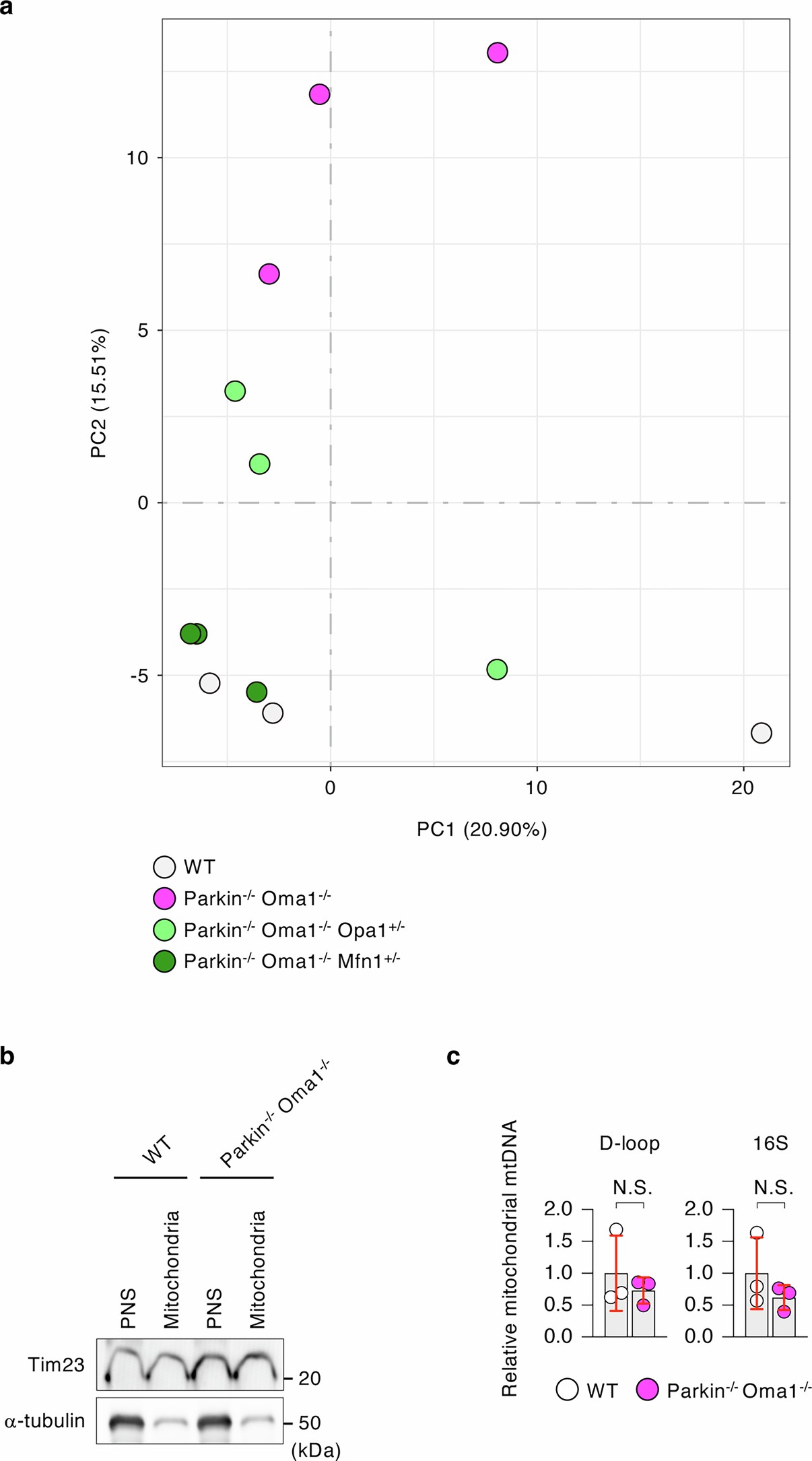
扩展数据图8 | 主成分分析和Western印迹分析
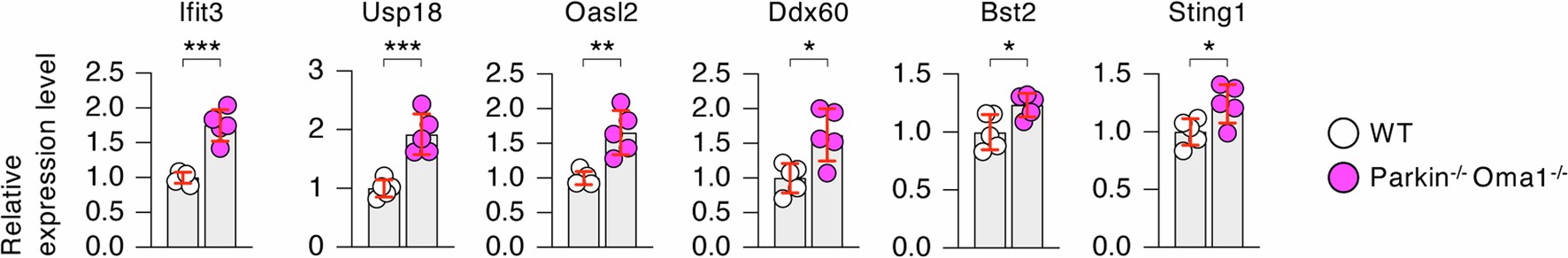
扩展数据图9 | qRT-PCR分析
6 .Parkin和OMA1在应激时限制线粒体的大小
线粒体分裂GTPase——DRP1,其缺失引起的线粒体增大导致MFN1的减少和OPA1裂解的增加,从而抑制肝细胞和其他细胞中过度的线粒体融合。由于Parkin-/-Oma1-/-小鼠的肝脏中没有形成巨线粒体(小编注:这里Parkin-/-Oma1-/-小鼠肝脏中没有形成巨线粒体的原因与DRP1没有关系,图4l和m中也展示出了Parkin-/-Oma1-/-小鼠肝脏中的DRP1水平并没有变化,Parkin和Omal敲除之后并不影响线粒体的自噬和泛素化(如图4n,o所示),因此线粒体自噬和泛素化会降解巨线粒体,所以并不会形成巨线粒体。该课题组之前发过一篇文章:Mitochondrial Safeguard: a stress response that offsets extreme fusion and protects respiratory function via flickering-induced Oma1 activation。研究人员在这篇文章中证明了p62可以介导非Parkin依赖的线粒体自噬过程中的线粒体泛素化,而且他们证明了巨线粒体是停滞的线粒体自噬过程中的中间体,而线粒体自噬会降解巨线粒体,保障体内线粒体稳态平衡。)(图2和扩展数据图4),因此研究人员检测了Parkin和OMA1是否在应对线粒体分裂应激时保护肝脏。研究人员利用floxed等位基因和肝细胞特异性Alb-Cre,构建了携带DRP1单敲除(Alb-Cre::Drp1flox/flox)、Parkin和OMA1双敲除(Alb-Cre::Parkinflox/floxOma1flox/flox)和DRP1、Parkin和OMA1三敲除(Alb-Cre::Drp1flox/floxParkinflox/floxOma1flox/flox)的肝脏特异性KO小鼠。这些KO小鼠体重正常(图4a);相比之下,三重KO小鼠与其他3个品系小鼠相比,表现出轻度的肝脏肥大(图4b)。与全身性的Parkin和OMA1双敲除小鼠一致(图2),通过激光共聚焦免疫荧光显微镜(图4c-e)和透射电子显微镜(图4f-k)分析,研究人员发现肝脏特异性的Parkin和OMA1 KO并不影响小鼠肝脏中的线粒体形态。正如预期的那样,DRP1KO增加了巨线粒体频率(图4d),线粒体大小(图4e,g),周长(图4h)和排列紊乱的嵴(图4i)(小编注:DRP1 KO会导致一定程度上的线粒体融合,使得线粒体增大,DRP1抑制OPA1/MFN1是一个机体保护线粒体过度融合而发生更严重后果的机制,类似负反馈机制。但是DRP1 KO本身仍会促进线粒体增大。)。此外,在DRP1KO小鼠肝脏中,线粒体失去了管状形态,纵横比降低(图4j),圆形度增加(图4k)。值得注意的是,在DRP1KO肝脏中额外缺失Parkin和OMA1进一步增加了巨线粒体频率(图4d),线粒体大小(图4e,g)和线粒体周长(图4h)。完整嵴的丢失也加剧(图4i)。
扩展阅读:DRP1缺失引起MFN1、OPA1水平降低机制
属于动力蛋白超家族的保守GTPase介导线粒体的分裂与融合。动态相关蛋白1(Dynamin-related protein 1,DRP1)与动态蛋白2(dynamin-2)可以收缩并切割线粒体,促进其分裂;而线粒体融合蛋白1和2( mitofusin,MFN1/2)以及视神经萎缩蛋白1会促进线粒体膜融合,使得线粒体的分裂与融合处于动态平衡状态。
OPA1在小鼠细胞中有五种亚型,包括两种长型(L1/L2)和三种短型(S3/S4/S5),线粒体内膜金属蛋白酶OMA1通过裂解L1和L2分别产生S3和S5亚型。
当DRP1缺失时,线粒体分裂水平降低,促使线粒体的尺寸增大,形成巨线粒体,由于线粒体过度融合会导致个体死亡,因此机体会出现一种保护机制,防止线粒体过度融合导致更巨大的伤害。当DRP1被敲除时,OMA1被线粒体膜电位闪烁(flickering,闪烁是指线粒体膜电位的快速、可逆的瞬时变化)激活,OMA1会将长型OPA(L-OPA1)1切割为没有活性的短片段(S-OPA1),此外,MFN1的表达水平也被限制,因此阻止了线粒体过度融合造成细胞或个体死亡。

OMA1切割OPA1模型图

保护线粒体过度融合的机制
参考文献:
[1] Murata D, et al. EMBO J. 2020.
[2] Saita S, et al. Genes Cells. 2016.
免疫印迹显示,DRP1缺失增加了OMA1介导的OPA1的裂解产物(S3和S5),减少了OMA1底物OPA1(L2)的形式,降低了MFN1和MFN2的水平(图4l,m),与先前报道的一致。Parkin和OMA1的额外缺失部分逆转了的这些变化(图4l,m)。这些数据表明,当线粒体分裂减少时,Parkin和OMA1阻止了线粒体的进一步扩大。
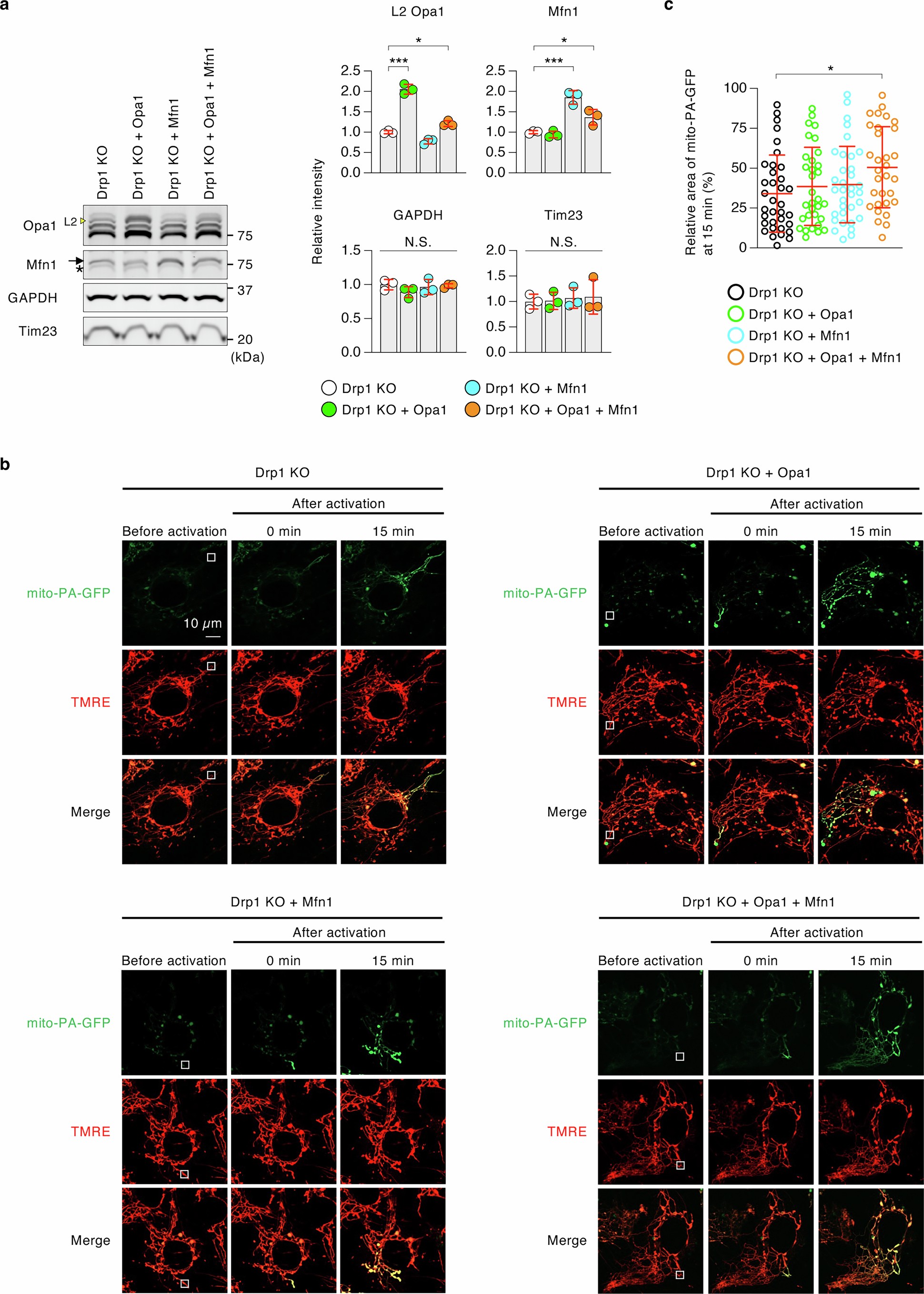
为了确定降低的OPA1-L2和MFN1水平在线粒体融合中的作用,因为在DRP1 KO小鼠中,胚胎成纤维细胞(mouse embryo fibroblasts,MEFs)与肝细胞中OPA1-L2与MFN1水平均下调,所以研究人员利用慢病毒转导系统在DRP1 KO小鼠的MEFs中恢复了这些蛋白水平。然后,研究人员使用基质靶向的光激活绿色荧光蛋白(matrix-targeted photoactivatable green fluorescent protein,mito-PA-GFP)来评估线粒体融合状态(小编注:PA-GFP是一种可以通过特定波长的光照激活的荧光蛋白。在未激活状态下,它不发出荧光;当用特定波长的光(如 405 nm 激光)照射后,它会转变为荧光状态,发出绿色荧光。通过将PA-GFP与线粒体基质靶向序列融合表达,可以将PA-GFP定位到线粒体基质中,形成mito-PA-GFP。在选定线粒体局部区域激活PA-GFP后,若线粒体发生融合,激活的荧光蛋白会因基质内容物混合而扩散至相邻线粒体。通过追踪荧光信号扩散的范围和速度,可定量评估线粒体融合活性(融合越活跃,荧光扩散范围越大)。线粒体融合后,内膜和基质形成连续结构,基质内容物(包括PA-GFP)可在融合的线粒体间自由交换。若观察到荧光信号从激活区域扩散至其他线粒体,则直接证明融合发生。反之,若荧光局限在初始激活区域,则表明融合被抑制)(扩展数据图10)。在本实验中,研究人员利用单个线粒体中mito-PA-GFP的光激活特性分析了每个线粒体的大小。由于线粒体分裂因DRP1的缺失而受到抑制,所以此研究分析测量的是单细胞中线粒体融合的水平。研究人员发现在DRP1 KO小鼠的 MEFs中,当OPA1和MFN1水平同时升高时,线粒体融合更多(扩展数据图10)。
在DRP1 KO小鼠肝脏中,线粒体的扩大降低了线粒体自噬,并导致泛素和p62在线粒体上的积累,p62是泛素连接酶复合物接头蛋白,又作为其亚基发挥作用。这些泛素化的、扩大的线粒体是停滞的线粒体自噬中间体,它们的泛素化需要p62,但不需要Parkin或PINK1。与这些研究一致,研究人员在DRP1 KO小鼠肝脏中发现了扩大的线粒体积累泛素和p62(图4n,o)。Parkin和OMA1的额外缺失进一步增加了三重敲除肝脏中这种停滞的线粒体自噬中间体,而Parkin和OMA1双重敲除的肝脏中则没有这种中间体(图4n,o)。为了直接检测线粒体自噬水平,研究人员通过尾静脉高压注射(小编注:这是一种将大体积质粒DNA溶液通过小鼠尾静脉注射进小鼠体内的方法。)将表达线粒体自噬生物传感器的质粒(即基质靶向的Su9-mCherry-GFP)(小编注:Su9来自线粒体靶向信号序列(MTS,mitochondrial targeting sequence),常来自酵母的线粒体ATP合酶亚基9(subunit 9),能把融合蛋白导入线粒体基质。这个质粒在转染后,表达的 Su9-mCherry-GFP 蛋白通过 Su9 信号导入线粒体基质,但它不会整合入线粒体DNA,也不是由线粒体自身表达的,是由细胞核转录后在细胞质中翻译,再导入线粒体的。)导入对照组、Alb-Drp1 KO、Alb-ParkinOma1 KO和Alb-Drp1ParkinOma1 KO小鼠的肝脏中(图4p,q)。最初,mCherry和GFP信号都在线粒体中观察到。由于线粒体通过线粒体自噬运输到溶酶体,而溶酶体的pH呈现酸性,最终只有mCherry信号仍然可以检测到(小编注:在正常情况下,Su9-mCherry-GFP 位于线粒体基质中,同时 GFP 和 mCherry 都有荧光(会看到黄色荧光,因为 GFP+RFP)。当线粒体进入自噬通路(mitophagy)时,整个线粒体被包裹进自噬小体,最终与溶酶体融合形成自噬溶酶体:溶酶体内部是酸性的。GFP 对酸敏感,荧光会被淬灭。mCherry 对酸不敏感,仍然发出红色荧光。所以黄色点(GFP+RFP):在线粒体基质中,正常未被降解的线粒体。红色点(RFP only):代表进入溶酶体的线粒体(线粒体自噬发生)。)。注射4天后,研究人员量化了肝脏切片中mCherry和GFP信号之间的比值,作为线粒体自噬通量(mitophagy index,线粒体自噬指数)的读数;线粒体自噬指数在Alb-Drp1 KO小鼠中显著降低,在Alb-Drp1ParkinOma1 KO小鼠中进一步加剧(图4p,q)。与此形成鲜明对比的是,Alb-ParkinOma1 KO小鼠中线粒体自噬并未减少(图4p,q)。这些数据表明,虽然Parkin和OMA1在肝脏中不参与线粒体泛素化,但它们在调节线粒体大小方面发挥着至关重要的作用,从而显著影响线粒体自噬
扩展阅读:线粒体大小与线粒体自噬
一句话总结:线粒体必须通过“分裂”变得更小,才能被有效地招募进入自噬系统,因此线粒体的大小(特别是过大的线粒体)是限制线粒体自噬发生的关键因素之一。
详细机制:
1. 线粒体必须变小才能被自噬体包裹
自噬体(autophagosome)有有限的包裹能力,直径约为 500–1500 nm。正常线粒体平均长度可能为 1–10 μm,尤其是高度融合状态下更长。因此,在启动线粒体自噬前,线粒体必须经过裂解/分裂(mitochondrial fission)变小。
2. Drp1 介导的线粒体分裂是自噬的前置步骤
Drp1 是主要的线粒体分裂蛋白。分裂能将线粒体分割为更小的片段,便于自噬体识别和包裹。实验观察显示,Drp1 缺失或抑制会阻碍线粒体自噬,因为线粒体无法变小进入自噬系统。
3. 过大的线粒体常常逃避自噬清除
融合态或肥大的线粒体(比如 Mfn1/2 过表达或 OPA1 促进融合)更不容易被识别为损伤对象。一方面因为其尺寸过大,另一方面可能因为融合后保留部分健康片段,整体信号不够“损伤”。
4. 线粒体大小与PINK1/Parkin通路关联
PINK1-Parkin 是细胞识别并清除“受损线粒体”的一套经典通路。健康线粒体有正常的膜电位。PINK1 被导入线粒体内膜,并被快速降解(如被 PARL 等蛋白剪切),不会积累。而受损线粒体的膜电位丧失,导致 PINK1 无法进入线粒体内膜,而在外膜累积。累积的 PINK1 招募并激活 Parkin。Parkin 泛素化多个线粒体外膜蛋白,标记线粒体进入自噬。即PINK1 在线粒体膜电位丧失时累积,招募 Parkin 启动线粒体自噬。长大或融合线粒体的膜电位恢复可能更强,延迟或抑制 PINK1 累积和 Parkin 招募。
该研究团队之前报道了线粒体增大诱导肝脏损伤,而通过基因敲除和敲低抑制线粒体融合来抑制巨线粒体可以改善肝脏中的这些损伤。与这些研究一致,Alb-Drp1 KO小鼠血清中肝酶丙氨酸氨基转移酶(ALT)释放增加(图4r)。研究人员发现Parkin和OMA1的额外缺失进一步升高了三重基因敲除小鼠的血清ALT水平(图4r)。这些数据表明,Parkin和OMA1共同保证了线粒体分裂缺陷导致的应激状态下线粒体的完整性和肝脏的健康。
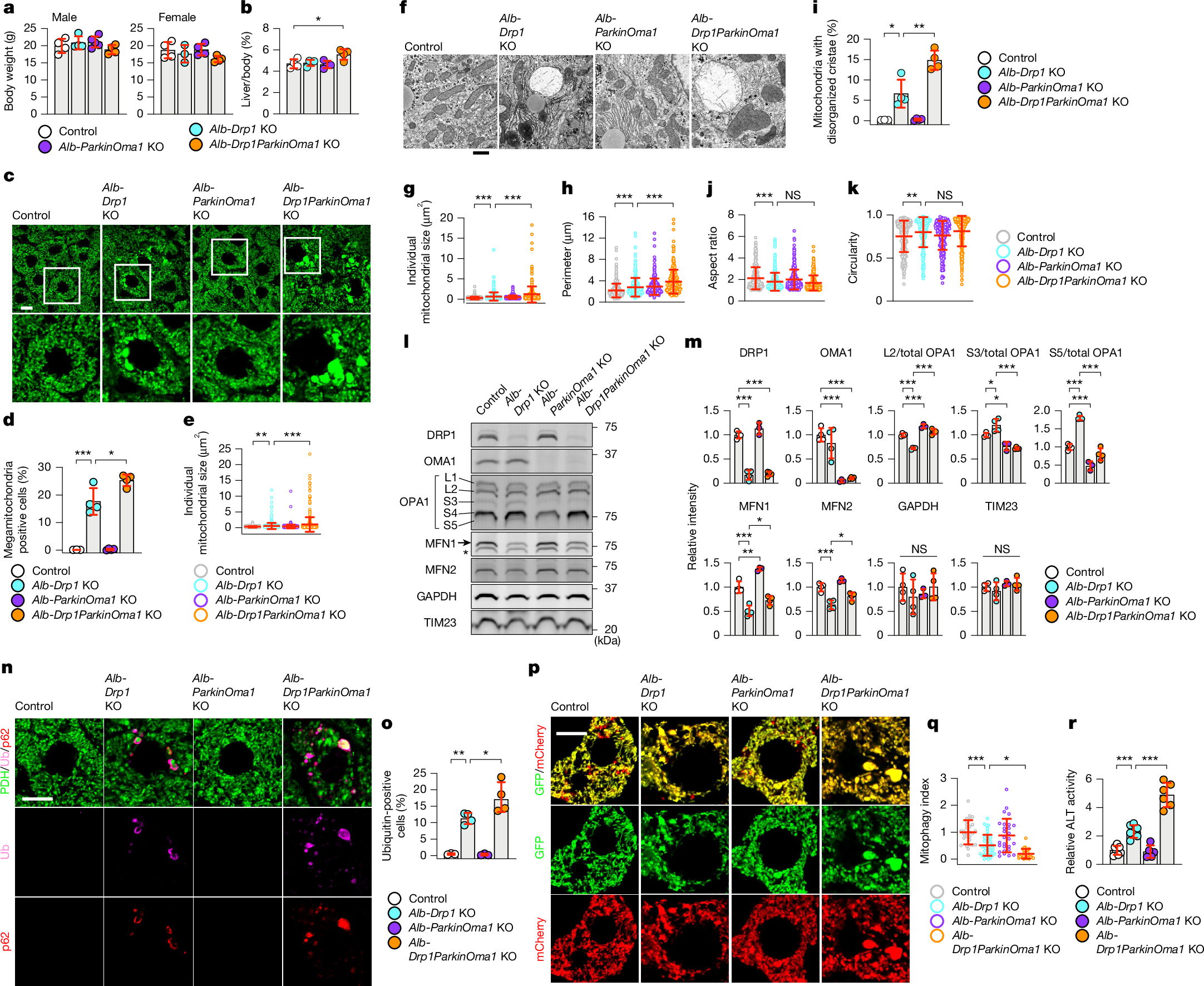
图4 | Parkin和OMA1抑制应激诱导的线粒体凋亡
扩展数据图10 | 线粒体融合状态的测量
7.Parkin和OMA1对运动神经元具有保护作用
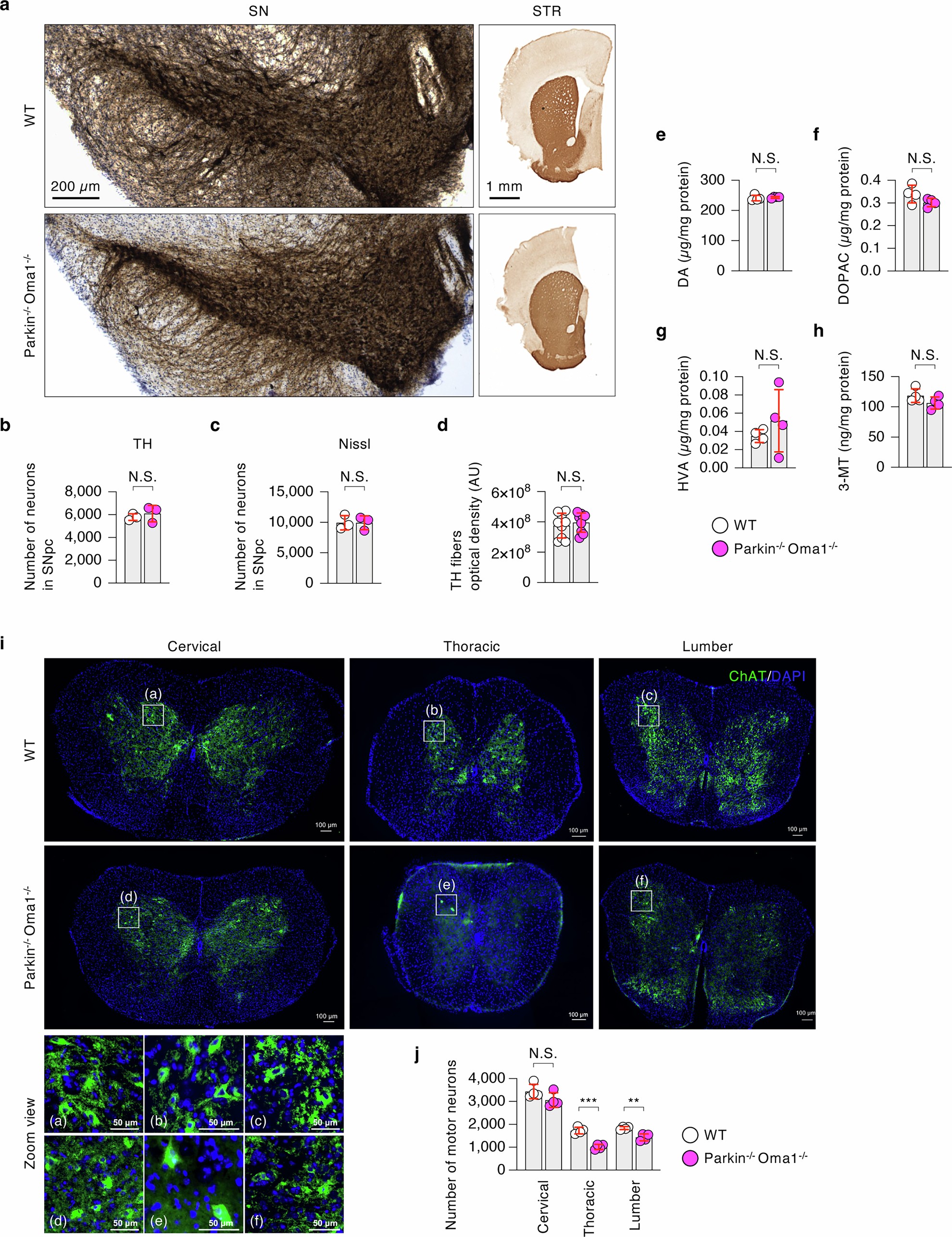
帕金森病与黑质多巴胺能神经元的丢失有关。Parkin-/-Oma1-/-小鼠的行为表型,包括活动减少和身体颤抖,可能是由于这些神经元的异常造成的(图1d)。此外,小鼠的双脚外翻和扭结的尾巴提示可能存在脊髓损伤。为了检测这两个区域,研究人员首先对WT和Parkin-/-Oma1-/-小鼠黑质致密部的酪氨酸羟化酶和Nissl阳性神经元进行了体视学计数(小编注: 尼氏染色液(Nissl Staining Solution)是神经生物学广泛使用的一种染色液,用于石蜡或冰冻切片神经元细胞浆中的尼氏小体(Nissl body)染色。被Nissl染色液染色的尼氏小体呈深蓝色,尼氏体是一种嗜碱性物质,分布广泛于各类神经元中,其形状、大小和数量在不同神经元中呈现差异。通过运用尼氏染色法,能够有效表征尼氏体,从而观察神经元内的细胞结构,并通过对尼氏体的观察来评估神经元是否受损。)。结果显示,两组小鼠的多巴胺能神经元数量无差异(扩展数据图11a-c)。研究人员还在WT和Parkin-/-Oma1-/-小鼠中发现,纹状体酪氨酸羟化酶阳性纤维密度水平相似(扩展数据图11a,d)。其次,研究人员检测了WT和Parkin-/-Oma1-/-小鼠大脑黑质部位多巴胺及其代谢物(3,4-二羟基苯乙酸、高香草酸和3-甲氧酪胺)的水平,发现两组小鼠水平相似(扩展数据图11e-h)。第三点,研究人员使用胆碱乙酰转移酶抗体在免疫荧光显微镜下评估了小鼠脊髓中的运动神经元,研究人员发现Parkin-/-Oma1-/-小鼠胸椎和腰椎区域运动神经元数量减少(扩展数据图11i,j)。这种脊髓损伤可能解释了Parkin-/-Oma1-/-小鼠行为和形态表型的改变。
扩展数据图11 | 多巴胺能神经元和运动神经元分析
总结
线粒体应激途径保护线粒体健康免受细胞损伤。然而,它们在生理条件下的作用在很大程度上是未知的。在本研究中,研究人员利用18种单、双和三重全身和组织特异性敲除和突变小鼠,结合系统的线粒体形态分析、非靶向代谢组学和RNA测序,发现了两个应激响应系统之间的协同作用——泛素E3连接酶Parkin和金属蛋白酶OMA1,通过外膜GTP酶MFN1和内膜GTP酶OPA1介导的线粒体融合来保护线粒体结构和基因组。虽然单独的Parkin或OMA1缺失不影响线粒体的完整性,但它们的联合缺失会导致小鼠体型变小,低运动活性,过早死亡,线粒体异常和先天免疫反应。因此,研究中的数据表明,Parkin和OMA1在线粒体外膜和内膜上维持着控制线粒体融合的双重调节机制,即使在没有外部压力的情况下也具有相同的功能。

小编注:这是本文研究工作模型。
(i)整个线粒体的融合反应需要外膜(outer membrane, OM)和内膜(inner membrane, IM)的融合。在WT小鼠中,线粒体融合受到外膜上的Parkin-PINK以及内膜上的OMA1调控,二者共同抑制了线粒体的过度融合。同时活化的OMA1会裂解DELE1,随后DELE1释放到细胞膜,引起ISR。
(ii)在Parkin−/−, Pink1−/− and Oma1−/−三种单基因敲除的小鼠中,其中调控线粒体融合的任意一种机制被抑制,还有另一种机制仍会抑制线粒体过度融合(如抑制OM上的调控机制,则IM上的调控机制仍可以阻断线粒体过度融合,反之同理)。
(iii)在Parkin−/−Oma1−/− 以及 Pink1−/−Oma1−/−小鼠中,抑制线粒体过度融合的两种机制都被阻断,因此线粒体会过度融合,从而导致小鼠过早死亡的现象。
(iv)在Parkin−/−Oma1−/− 以及 Pink1−/−Oma1−/−小鼠中引入OPA1或MFN1的杂合敲除可以模仿调控线粒体融合其中一种机制被抑制,而另一种功能进行补偿调控的机制来防止线粒体的过度融合,与(ii)中机制类似。
原文链接:https://www.nature.com/articles/s41586-025-08590-2IF: 50.5 Q1
https://wap.sciencenet.cn/blog-3483272-1481399.html
上一篇:代谢学人——Nature Cell Biology:PPID,燃脂减肥“新钥匙”
下一篇:Science:糖尿病,“细胞身份丢失”引起的全身崩溃