博文
代谢学人——Nature Cell Biology:PPID,燃脂减肥“新钥匙”
||
撰文 | 李欣茜 郑宇含 张彦康 张婷 仲银召 李雨
编辑 | 孟美瑶
校对 | 张婷

研究背景
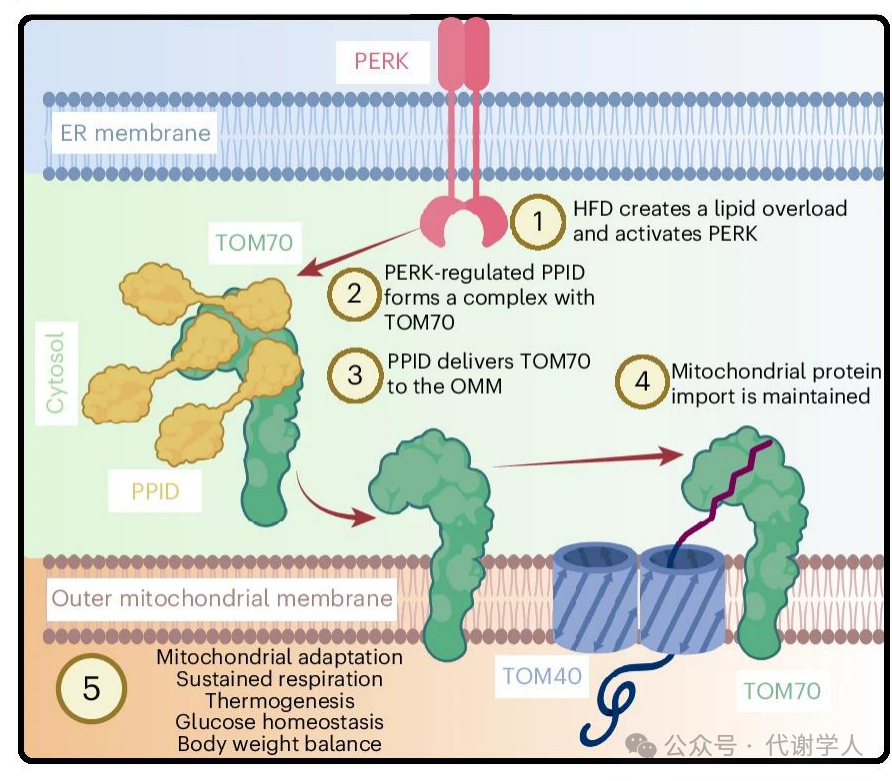
肥胖是一种非常普遍的临床前症状,也是多种慢性疾病如2型糖尿病、心血管疾病和癌症的危险因素。在当今世界,包括成年人和儿童在内已有大约40%的人超重,13%的人肥胖。由于遗传因素、患者不依从性或治疗成本过高,运动、限制饮食以及减肥手术等肥胖治疗的效果甚微。许多研究表明,BAT(棕色脂肪组织)在寒冷刺激或运动条件下,可被激活并启动产热程序,通过氧化分解代谢促进机体能量消耗,增强胰岛素敏感性,因此,激活BAT可能是治疗肥胖及其代谢性疾病的一个有效治疗策略。
BAT的产热功能依赖于线粒体与其他细胞器(如内质网)之间的相互作用。细胞器间通讯的减少或失败可促进肥胖、2型糖尿病等代谢疾病的发展。OMM(线粒体外膜系统)是线粒体与其他细胞器交流的重要结构,其上的蛋白复合体控制着线粒体蛋白或脂质的输入过程,是维持线粒体功能的基础,但这些蛋白复合体如何定位在OMM上尚不清楚。内质网为蛋白质-蛋白质相互作用、信号传导和运输搭建桥梁,能快速响应饮食诱导的应激,维持适应性细胞功能。在内质网中,PERK(蛋白激酶R(PKR)样ER激酶)控制线粒体脂质和蛋白质导入,促进线粒体呼吸作用,使内质网信号与能量和代谢相适应(小编注:蛋白进入线粒体主要依赖于线粒体靶向序列,由线粒体膜TOM/TIM复合体介导转运,并不直接依赖于内质网。而内质网调控线粒体蛋白的导入,主要通过PERK下游信号(如ATF)调控线粒体蛋白的表达水平,或通过调控线粒体外膜蛋白(如TOM70)活性或定位促进线粒体蛋白的导入)。目前研究发现,高脂肪饮食(HFD)会诱发BAT中的内质网应激,并激活产热以维持机体能量平衡和胰岛素敏感性。然而,在应激期间,维持细胞能量代谢的细胞器间通讯机制,特别是内质网向OMM发出信号和调节OMM蛋白定位的调控机制,仍不清楚。
在本篇文章中,研究人员发现PERK缺失导致PPID(胞质分子伴侣蛋白)蛋白水平下降,进而抑制TOM70在OMM上的导入和线粒体功能,导致肥胖、葡萄糖不耐受和胰岛素抵抗。进一步研究发现PPID通过其TPRs(四肽重复序列)和PPIase(肽基辅氨酰异构酶)结构域活性来促进TOM70的OMM导入,从而调节棕色脂肪细胞的线粒体呼吸和产热功能。总之,这些发现揭示了PPID在维持线粒体功能和代谢健康中的关键作用,对治疗肥胖和2型糖尿病具有重要意义。
拓展阅读:PERK调控线粒体蛋白质和脂质导入
[1] Balsa E, et al. Mol Cell. 2019. [2] Latorre-Muro P, et al. Cell Metab. 2021. [3] Sassano ML, et al. J Cell Biol. 2023. [4] Perea V, et al. EMBO J. 2023.在内质网应激状态下,内质网中错误折叠或未折叠蛋白的积累可触发UPR(未折叠蛋白反应),激活PERK,以PERK-eIF2α-ATF4通路诱导SCAF1(超复合物组装因子1)表达,促进线粒体呼吸链超复合物形成,增强线粒体呼吸;也有研究发现冷刺激激活棕色脂肪细胞PERK活性,促使OGT(O-GlcNAc转移酶)磷酸化,磷酸化的OGT诱导TOM70发生糖基化修饰,增强MIC19蛋白进入线粒体,进而促进线粒体嵴形成和呼吸作用。此外,有研究发现PERK可与EMCS(内质网-线粒体接触位点)上的E-Syt1(脂质转移蛋白)相互作用,维持内质网和线粒体之间磷脂的运输以及线粒体呼吸作用;也有研究发现PERK活性可促进线粒体膜PA(磷脂酸)的适应性重塑,诱导线粒体伸长以适应内质网应激。
拓展阅读:PPID蛋白
[1] Dunyak BM, et al. J Med Chem. 2016. [2] Dimogkioka AR, et al. RSC Adv. 2021.PPID(也称为CYP40)是肽基脯氨酸异构酶家族的成员之一,含有PPIases结构域,可催化脯氨酸的顺式/反式异构化,这在多种蛋白质的折叠、激活或降解过程中发挥调控开关作用,这些蛋白包括了在癌症、神经退行性疾病等中发挥关键作用的蛋白,表明PPIase可能是治疗疾病中的重要靶点,但PPIase活性位点较浅,且该家族成员之间具有很好的保守性,使特异性药物的设计具有挑战性。此外,PPID是此家族中唯一含有TPR结构域的蛋白,可与HSP90结合,促进未活化的类固醇受体折叠形成高亲和力的类固醇结合状态。本篇文章发现PPID在棕色脂肪组织中发挥着重要作用,通过TPR结构域与TOM70相互作用,并通过PPIase结构域帮助其正确导入线粒体外膜,从而调控脂肪细胞的能量代谢。

敲黑板啦!
1.PERK缺失影响TOM70导入OMM和线粒体功能,导致小鼠代谢紊乱
2.PERK依赖性PPID协助TOM70导入OMM,进而调控线粒体蛋白的导入
3.PPID通过TPR和PPIase结构域与TOM70特异性结合,促进TOM70导入OMM
4.PPID缺失导致小鼠代谢紊乱
研究结果
1.PERK缺失导致TOM70无法有效导入OMM
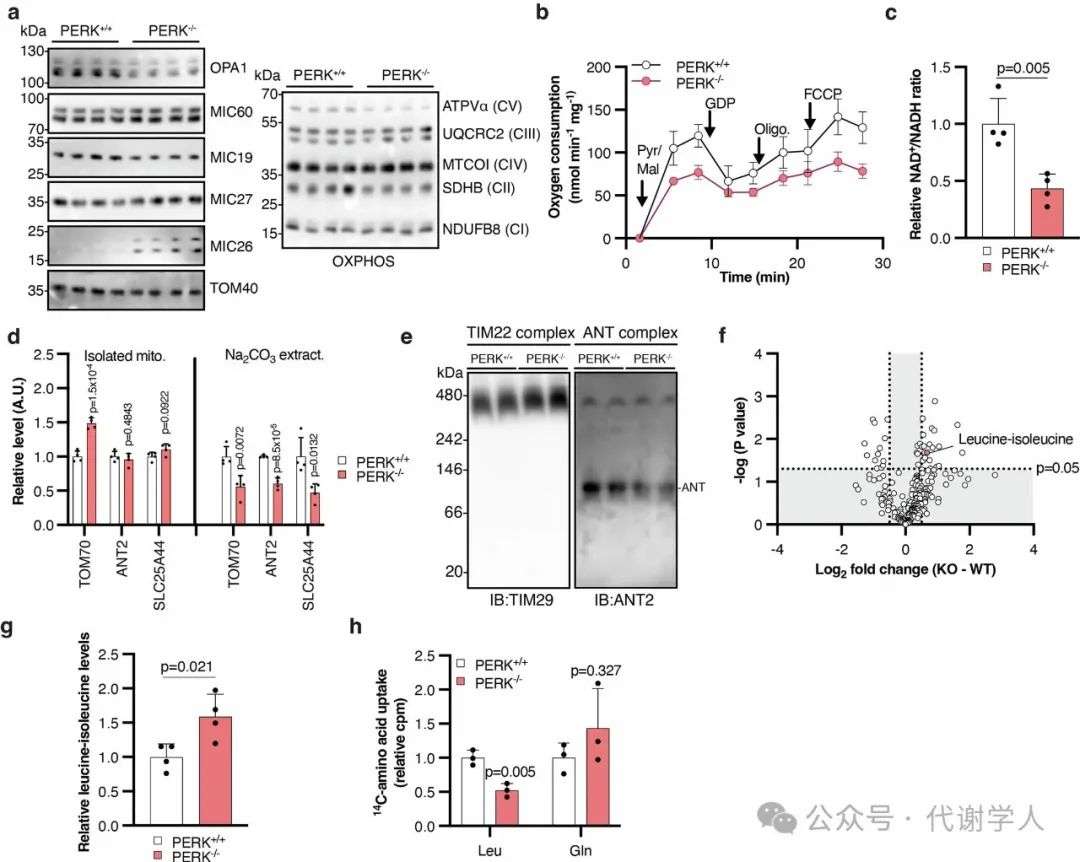
棕色脂肪细胞含有大量线粒体,这些线粒体具有密集的嵴,能在营养或脂肪酸过剩的情况下调控产热功能、葡萄糖稳态和体重。如前所述,棕色脂肪细胞的调节功能依赖于线粒体与内质网(ER)等细胞器之间的通讯,ER中的PERK控制线粒体脂质和蛋白质向内运输,促进活性嵴形成和耗氧量增加,从而调控能量代谢。为了探究PERK调控呼吸和产热的分子机制,研究人员构建了脂肪细胞特异性(AdipoQ-Cre)或棕色脂肪细胞特异性(UCP1-Cre)PERK缺失(PERK-/-)小鼠模型。HFD喂养且长期热中性(30℃)条件下,与PERK+/+小鼠相比,两种PERK-/-小鼠体重均显著增加(图1a和扩展数据图1a、b)。同时,脂肪特异性缺失PERK小鼠的耗氧率降低(图1b),摄食量没有明显差异(扩展数据图1c),这表明PERK缺失引发的肥胖是由棕色脂肪细胞产热功能缺陷引起的。另外,脂肪特异性缺失PERK小鼠表现出冷不耐受(扩展数据图1d)和胰岛素抵抗(图1d和扩展数据图1g),两种PERK-/-小鼠均表现出葡萄糖耐受不良(图1c和扩展数据图1e-g)和空腹血糖水平升高(图1d和扩展数据图1f)。总之,研究人员发现,产热脂肪细胞中PERK的缺失会导致全身能量代谢失衡,从而导致饮食诱导的肥胖、冷不耐受、葡萄糖不耐受和胰岛素抵抗。
接下来,研究人员对线粒体结构及呼吸链功能进行检测。电镜结果显示,HFD喂养的PERK-/-小鼠肩胛间棕色脂肪组织(iBAT)中线粒体嵴密度降低(图2a和扩展数据图2)。同时PERK-/-小鼠iBAT线粒体中MIC19(线粒体连接位点和嵴形成系统(MICOS)的重要亚基,能调控线粒体嵴结构)和OPA1(一种线粒体融合蛋白,参与线粒体形态维持和嵴形成)蛋白表达明显抑制(小编注:MIC19、MIC60、MIC27和MIC26都是MICOS(线粒体连接位点和嵴形成系统)复合体中的亚基,能调控线粒体嵴结构和功能,但具体作用存在差异。MICOS主要分为两个亚基,MIC60-MIC19-MIC25亚基主要负责在线粒体内外膜之间形成连接位点,并维持嵴连接结构,以保证线粒体结构的完整性。其中MIC19能促进2个MICOS亚复合物相互作用,维持MICOS复合物的完整性和功能。同时MIC19还能介导线粒体外膜和内膜接触,并促进内膜折叠形成嵴。MIC60能够独立于其他MICOS复合体成员在线粒体内膜中组装,表明它可能具有自我组装的能力,并且MIC60能在线粒体内膜中形成离散的焦点结构,这些结构可能是新生的嵴连接点。MICOS另一个亚基是MIC10-MIC26-MIC27亚基,主要负责嵴的形成并稳定嵴连接结构。MIC26和MIC27的稳态水平能相互调节,其中一种的敲除或过表达总是伴随着另一种蛋白质的增加或减少,其表达高低对线粒体嵴造成的影响尚未明晰)。此外,研究人员发现与PERK+/+小鼠相比,从PERK-/-小鼠iBAT中提取的线粒体的呼吸能力降低(图2b和扩展数据图3b)(小编注:扩展数据图3b中,首先加了丙酮酸和苹果酸(pyruvate+malete)混合物,这两种化合物进入线粒体后,分别在丙酮酸脱氢酶或苹果酸脱氢酶的作用下产生NADH进入电子传递链,产生电子流和质子梯度,驱动ATP合成和氧气的利用,OCR水平升高;随后加GDP,GDP能与解偶联蛋白(UCPs)结合并抑制其功能,抑制线粒体内膜的质子漏。UCPs会导致质子梯度耗散,使OCR水平升高而不生成ATP,加入GDP可以抑制这种非产能性耗氧,从而评估在没有UCPs介导的解偶联下的呼吸速率;此外,在本文中,研究人员利用添加pyruvate+malete和添加GDP后的耗氧量计算UCP1活性(图2b和图7d),具体公式为O2UCP1=O2pyr/mal-O2GDP。随后加oligomycin的作用是抑制ATP合酶,阻断氧化磷酸化,正常情况下会抑制OCR水平,但是这里作者没有解释为什么加了Oligomycin后曲线上升,推测可能的原因是GDP对UCPs的抑制作用逐渐消退,质子漏增加,促进OCR水平;最后加FCCP会破坏质子梯度和线粒体膜电位,使电子传递链以最大速率运),NAD+/NADH比例下降(扩展数据图3c)。总之,这些结果表明,PERK缺失会破坏线粒体结构,导致线粒体呼吸和产热功能障碍,进而诱发肥胖和葡萄糖稳态失调。
据报道,PERK能调控线粒体蛋白向内运输,从而正向调控线粒体嵴结构和呼吸功能,该过程主要由TOM70信号通路介导。TOM70和TOM20是线粒体外膜(OMM)上两个重要的线粒体导入受体,能特异性识别并将蛋白转运到线粒体内膜中发挥作用。例如,TOM70介导腺嘌呤核苷酸转运酶2(ANT2)导入,TOM20介导鸟氨酸氨基甲酰转移酶(OTC)导入。于是研究人员提取HFD PERK-/-小鼠iBAT中的线粒体,分别加入35S-ANT2和35S-OTC,并检测二者向线粒体内转运的效率。结果显示,35S-ANT2无法正常转运,而35S-OTC不受影响(图2c、d)(小编注:2c中,作者通过添加10μM FCCP使线粒体膜对质子完全通透,消除膜电势。有研究表明,蛋白质导入线粒体的过程需要膜电势。经典的线粒体蛋白质导入途径是前导肽途径。这些蛋白带有带正电荷的前导肽,内膜两侧的膜电位(在基质一侧为负)对带正电荷的前导肽产生电泳作用,推动前体蛋白转位到基质中。带前导肽的内膜蛋白在经历剪切后,会有precursor转变为mature蛋白。不带前导肽的内膜蛋白通过膜间的小TIM伴侣和内膜的载体转位酶(如TIM22)导入内膜,膜电势能激活TIM22等载体蛋白,从而促进蛋白导入线粒体内膜。因此,线粒体膜电势的大小是内膜蛋白易位效率的关键决定因素。因此这里用FCCP破坏膜电势后,导入的蛋白变少了。加蛋白酶K是为了去除未导入线粒体内的35S-ANT2和35S-OTC蛋白,减少假阳性。同样的,后文检测线粒体外膜蛋白TOM70导入情况时未使用蛋白酶K处理,而用Na2CO3缓冲液洗去结合松散的TOM70,这是因为TOM70是线粒体外膜蛋白,若使用蛋白酶K则会影响已经导入的TOM70蛋白的检测),这表明PERK缺失导致TOM70信号通路受损。然而,与此矛盾的是,HFD PERK-/-小鼠iBAT分离的线粒体中TOM70水平反而较高(图2e和扩展数据图3d)。于是,研究人员用pH为11.5的Na2CO3缓冲液洗去线粒体表面的非紧密结合蛋白,发现PERK-/-显著抑制了TOM70。ANT2和SLC25A44(支链氨基酸(BCAA)转运蛋白同样依赖于TOM70信号通路向内转运,其能维持棕色脂肪细胞的高氧化率和能量代谢)的表达水平(图2e和扩展数据图3d)。研究人员还在提取的线粒体中加入35S-TOM70,发现HFD PERK-/-的线粒体中35S-TOM70的水平降低(图2f)。以上结果表明,PERK缺失后TOM70无法有效导入OMM中发挥转运功能。
据报道,TOM70通路通过运输具有内部线粒体靶向序列(MTS)的未折叠的线粒体载体蛋白和蛋白质以维持线粒体的生物能量学功能。接下来,研究人员对TOM70转运功能缺陷导致的能量代谢异常进行进一步探究。PERK缺失导致转运到线粒体内膜(IMM)上的ANT2减少(图2c和扩展数据图3e),由于ANT2主要负责腺嘌呤核苷酸的转运,帮助维持线粒体内外的ATP/ADP平衡,在氧化磷酸化过程中发挥关键作用,其缺陷会抑制线粒体的氧化能力,抑制产热,这与HFD PERK-/-小鼠冷不耐受表型相符(扩展数据图1d)。有研究表明,SLC25A44功能障碍会导致支链氨基酸无法有效地被转运到线粒体基质中,从而降低线粒体氧化代谢效率。根据代谢组学分析,研究人员发现HFD PERK-/-小鼠iBAT中亮氨酸、异亮氨酸水平显著升高(扩展数据图3f、g)(小编注:据报道,BAT对于降低全身BCAA水平具有重要作用。SLC25A44在棕色脂肪细胞中表达较多,能促进亮氨酸等运输到线粒体中,从而被氧化清除。敲除SLC25A44后,棕色脂肪细胞中BCAA的氧化速率显著降低。在这里,研究人员做的是BAT的代谢组学分析,检测到的是BAT中BCAA增多,而非线粒体BCAA增多。可能是因为PERK敲除后,抑制SLC25A44导入,会抑制BCAA转入线粒体中被氧化消耗,从而造成整个BAT组织中BCAA增多)。此外,体外14C氨基酸摄取实验结果显示,HFD PERK-/-小鼠线粒体中转入的亮氨酸减少,表明其向内转运功能受损,而谷氨酰胺转入水平不变(图2g和扩展数据图3h)。总之,这些结果表明,PERK缺失导致TOM70无法有效导入OMM,使得线粒体功能相关蛋白质无法向内转运,进而导致iBAT中线粒体产热和能量代谢功能异常。
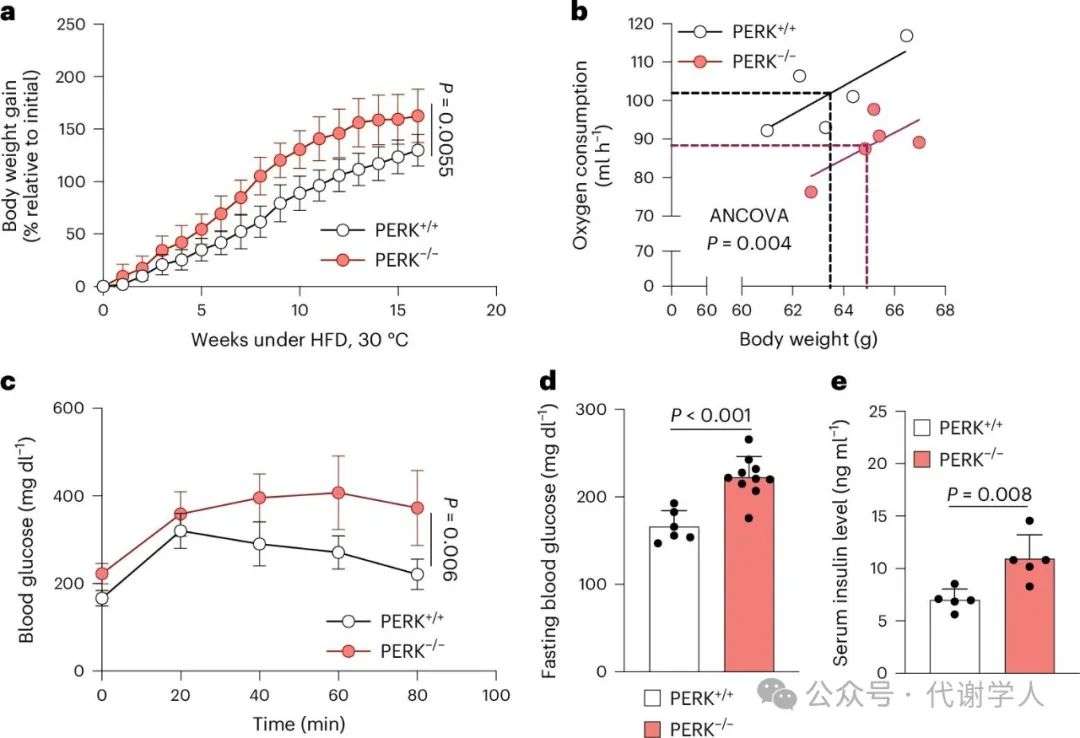
图1.PERK缺失导致肥胖和葡萄糖稳态受损
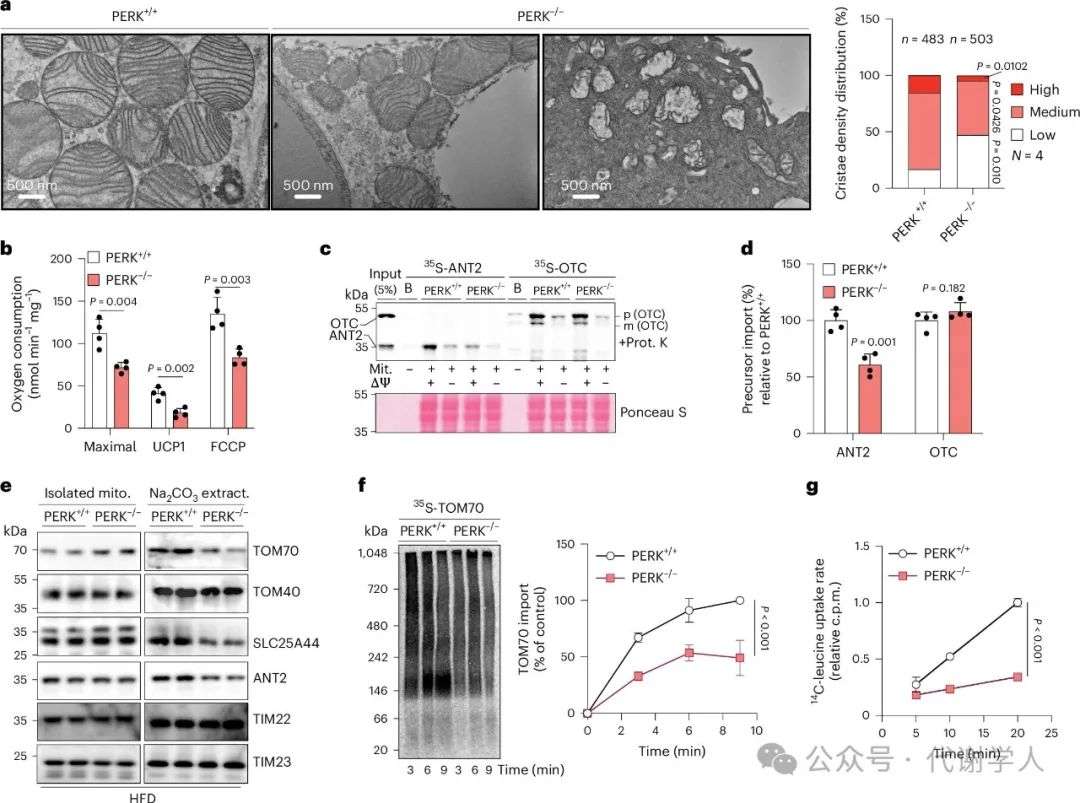
图2.PERK缺失抑制TOM70导入和线粒体功能
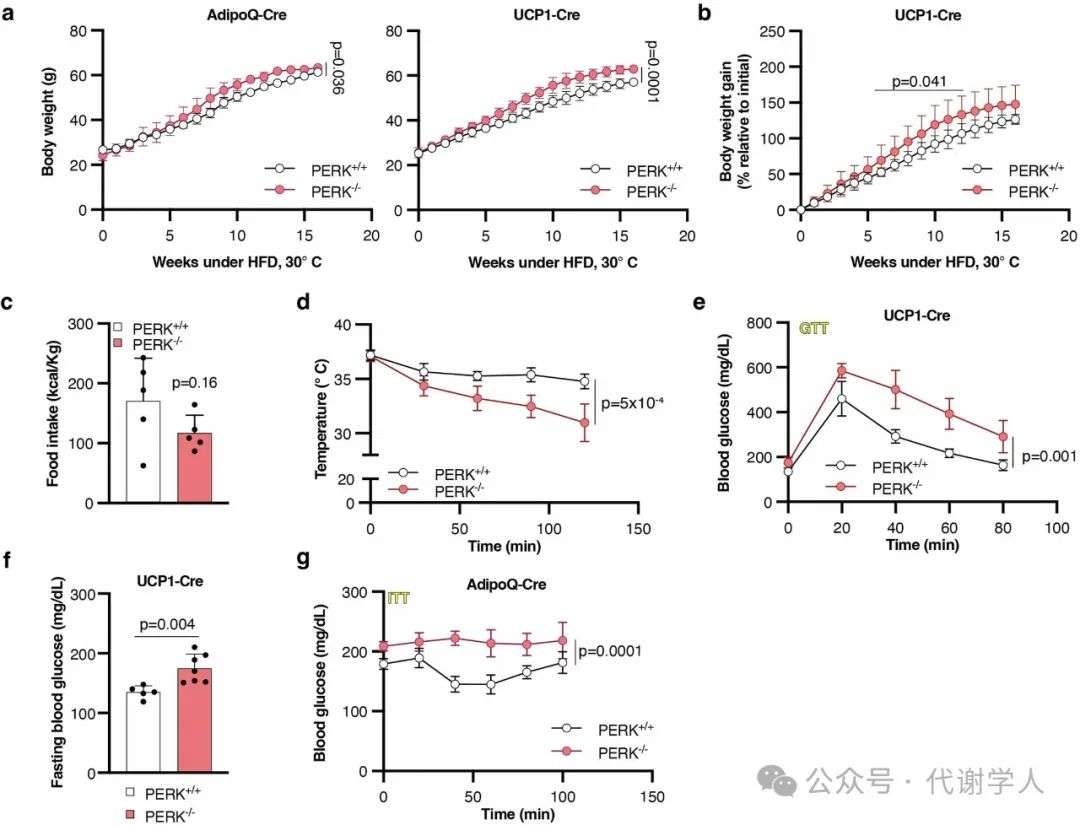
扩展数据图1.HFD PERK-/-小鼠的生理参数
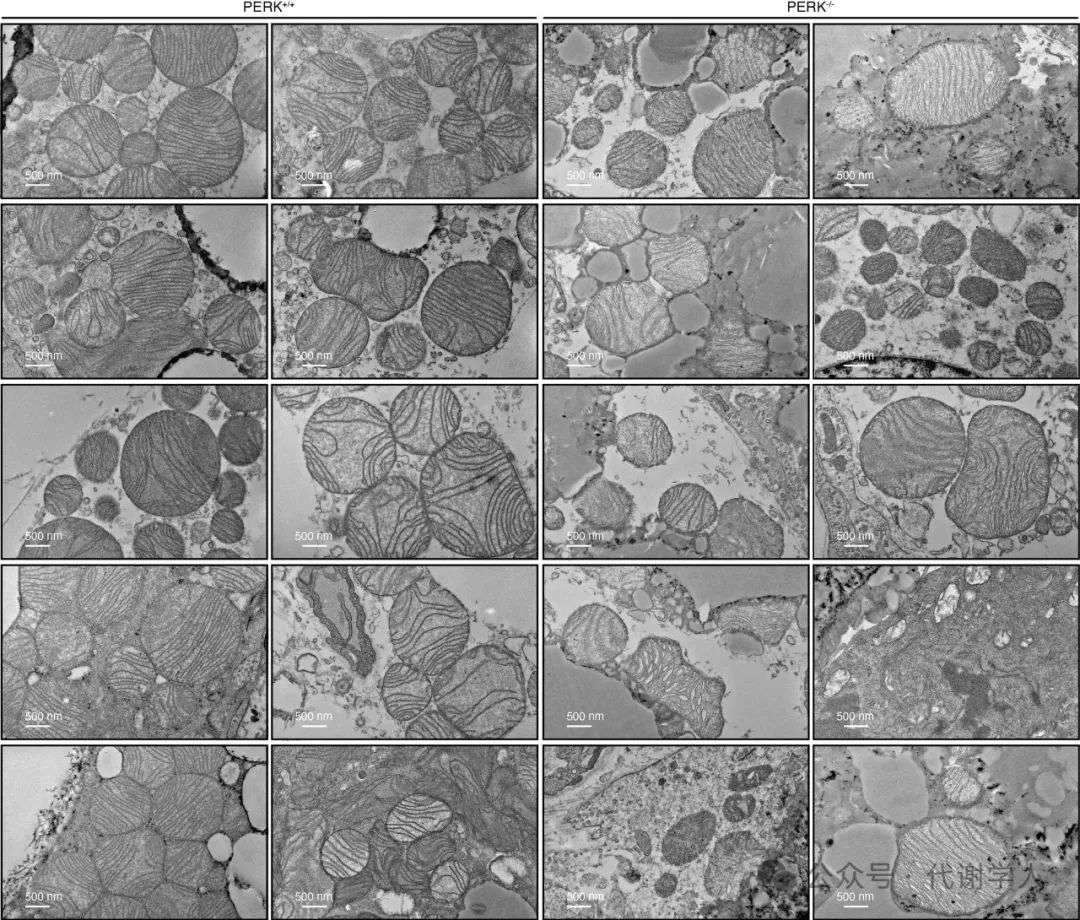
扩展数据图2.电镜下HFD PERK-/-小鼠iBAT中分离的线粒体的超微结构
扩展数据图3.在HFD条件下,PERK缺失影响线粒体蛋白
2.PPID调控TOM70导入和呼吸作用
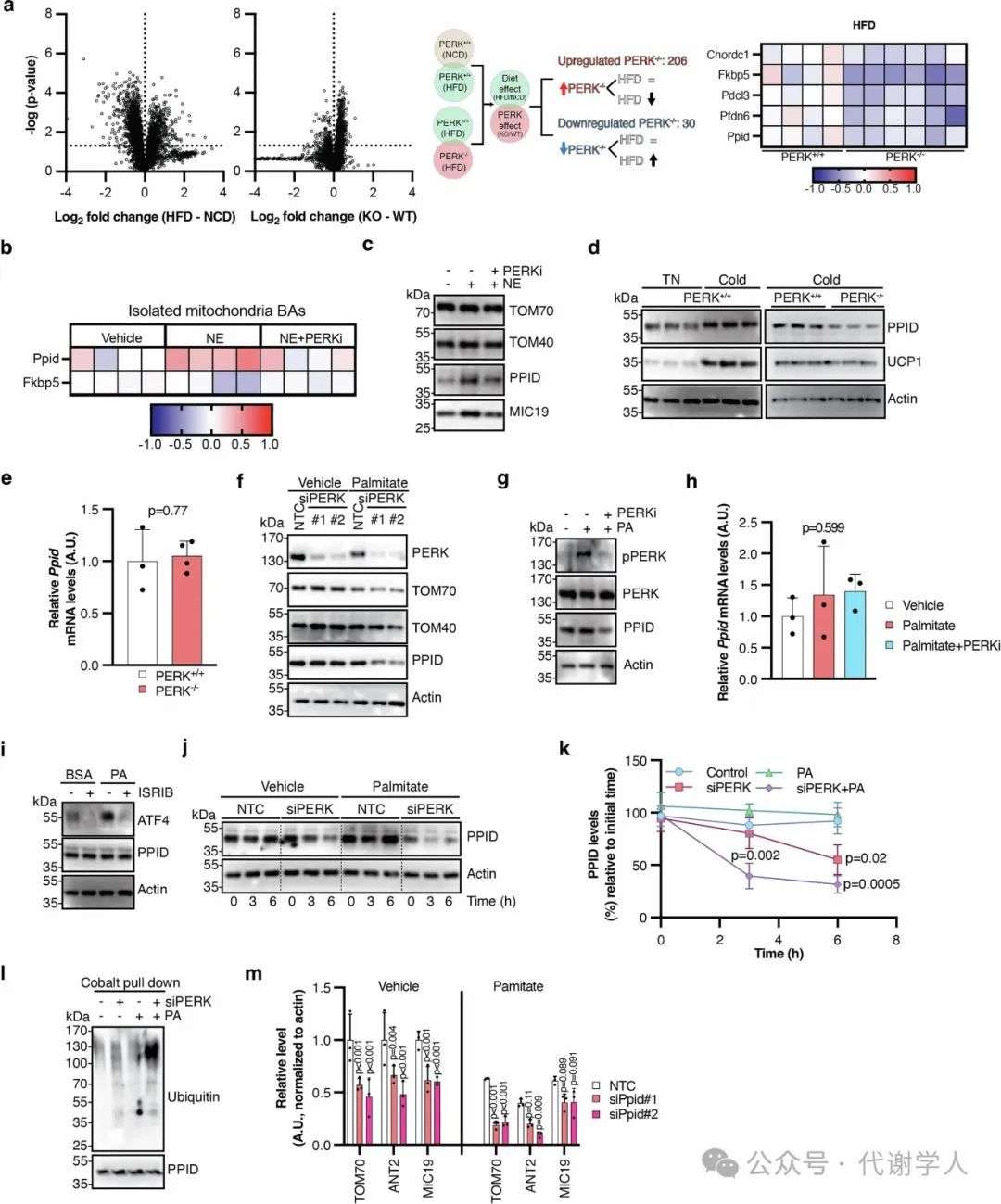
为了探究饮食诱导的肥胖下,PERK依赖性TOM70导入OMM的机制,研究人员首先寻找调控TOM70导入OMM的蛋白。研究人员分别将NCD与HFD、HFD PERK+/+与HFD PERK-/-小鼠的iBAT进行蛋白质组学分析,在两组蛋白质组学数据集中共同检测到的蛋白质有5036种,根据q<0.05筛选出受PERK调控而不受高脂饮食影响的基因,其中PERK缺失导致206个蛋白质上调,30个蛋白质下调(扩展数据图4a)。蛋白质网络和GO分析表明,有57%(119/206)的上调蛋白富集到了线粒体相关通路(图3a,b和扩展数据图4a),同时,细胞质蛋白有所减少,特别是具有辅助蛋白折叠功能的分子伴侣蛋白表达下调,包括CHORDC1,FKBP5,PDCL3,PFDN6和PPID(扩展数据图4a)。研究人员对从HFD小鼠iBAT中分离的线粒体进行蛋白质组学分析,并着重分析了图3b的分子伴侣蛋白簇中的蛋白,结果表明PERK缺失显著抑制PPID(图3c)。在此前的质谱数据集中,研究人员在棕色脂肪细胞提取的线粒体中只检测到了FKBP5和PPID两种细胞质伴侣蛋白受PERK调控,并且只有PPID显示出PERK依赖性(扩展数据图4b,c)。随后,研究人员发现冷刺激促进PPID蛋白表达,而PERK缺失显著抑制其表达(扩展数据图4d)。总之,以上结果表明,PERK缺失会抑制细胞质中分子伴侣蛋白PPID表达。
PPID属于肽基脯氨酰异构酶家族,该家族包含PPIA-PPIH共八种蛋白,而PPID是其中唯一含有四肽重复序列(TPR)的蛋白,TPR可以识别底物以调控蛋白质的折叠和组装。接下来,研究人员进一步探究了PPID是否调控TOM70和线粒体功能。WB和基因表达结果显示,HFD PERK-/-小鼠iBAT中PPID蛋白水平下调,但mRNA表达水平没有变化(图3d和扩展数据图4e)。同样,在棕色脂肪细胞中,用棕榈酸(PA)处理模拟高脂饮食的代谢压力,敲减或药物抑制PERK均能显著抑制PPID蛋白水平,但不影响mRNA表达水平(扩展数据图4f-h)。值得注意的是,体外敲减PERK后,PPID蛋白水平的抑制伴随着TOM70蛋白水平的下调(图3e和扩展数据图4f),而过表达PPID能显著促进TOM70蛋白水平(图3e)。此外,在棕色脂肪细胞中过表达PPID能上调细胞呼吸水平(图3f)。这些结果表明,PPID能调节TOM70蛋白表达,进而调控线粒体呼吸功能。
据报道,真核起始因子2α-激活转录因子4(eIF2α-ATF4)途径是控制蛋白质翻译的经典信号通路,在内质网应激、氧化应激等条件下,细胞内调控翻译的eIF2α会被磷酸化,从而促进ATF4翻译,随后ATF4结合到下游基因的启动子区域,促进分子伴侣蛋白的表达,帮助未折叠或错误折叠的蛋白质正确折叠。于是,为了探究PERK是否通过elF2α-ATF4途径来调控PPID水平,研究人员使用整合应激反应抑制剂(ISRIB)(据报道能抑制PERK-elf2α信号通路)体外处理棕色脂肪细胞,发现ISRIB能显著抑制ATF4,但不影响PPID表达水平(扩展数据图4i),表明PERK对PPID的调控不依赖于经典的elF2α-ATF4途径。由于PERK不影响PPID的mRNA表达水平(扩展数据图4e,h),于是研究人员利用环己酰胺追踪实验等从蛋白质层面进一步探究PERK对PPID的调控机制。环己酰胺能抑制蛋白质的合成,常用于研究细胞内蛋白质半衰期、蛋白质降解等。在添加环己酰胺后不同时间点对胞内蛋白进行分析,研究人员发现敲减PERK抑制胞内PPID水平,促进蛋白质泛素化修饰,PA处理进一步加剧这种影响(扩展数据图4j-l),表明PERK可能通过泛素化修饰对PPID进行翻译后调控。总之,抑制PERK能下调细胞质伴侣蛋白PPID水平,同时伴随着TOM70水平和呼吸能力的下降。当增加PPID的表达时补偿PERK抑制带来的影响,恢复TOM70水平和细胞呼吸,表明PPID在调控线粒体功能中起重要作用。
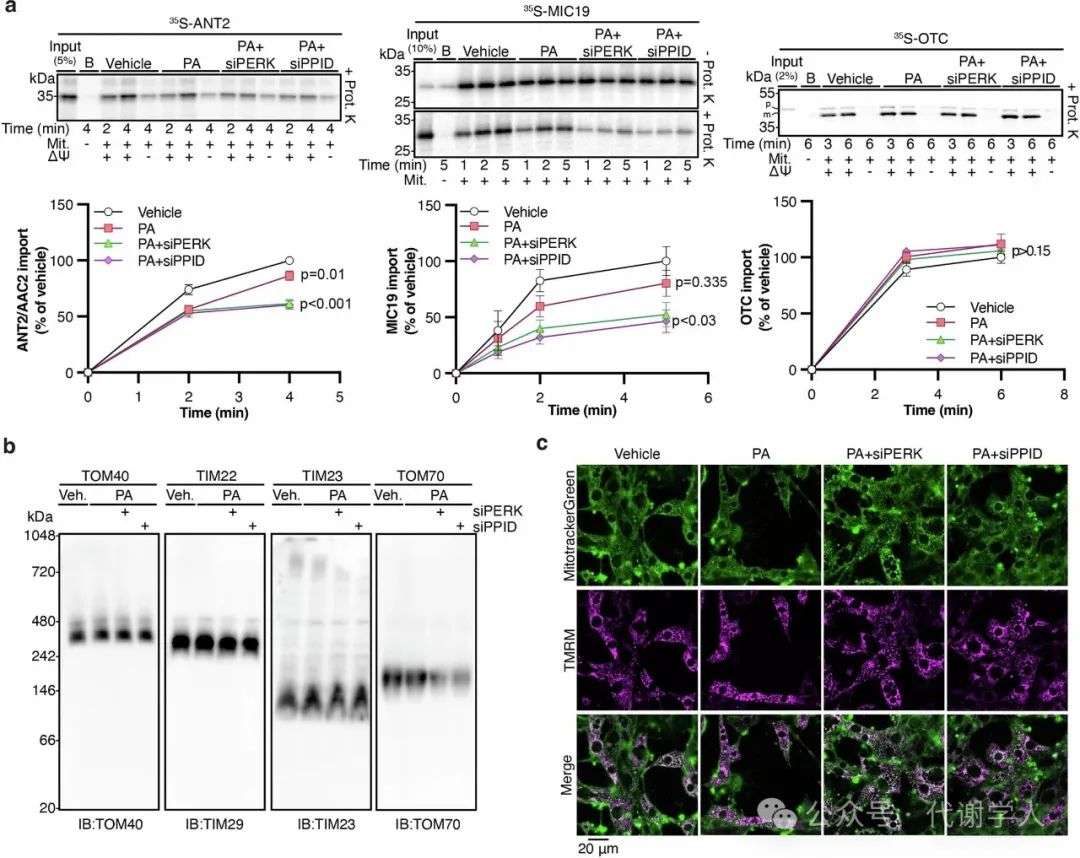
接下来,为了评估PPID调节TOM70导入OMM的能力,研究人员提取小鼠iBAT线粒体进行了体外导入实验。结果表明,外源添加重组PPID能提高35S-TOM70的线粒体掺入率(图3g)。同样,与对照组相比,来自敲减Ppid的棕色脂肪细胞的线粒体中35S-TOM70的体外导入减少(图3i和扩展数据图4m),并伴随着TOM70依赖性底物35S-MIC19和35S-ANT2导入水平降低,而TOM20依赖性底物35S-OTC不受影响(扩展数据图5a)。与此一致,WB结果显示敲减Ppid显著抑制了棕色脂肪细胞中TOM70、ANT2和MIC19的蛋白水平,且在PA处理下差异更加显著(图3h和扩展数据图4m)。与敲减Ppid类似,在敲减PERK的棕色脂肪细胞(扩展数据图5a)和PERK-/-小鼠iBAT(图2c、d)的线粒体中也观察到TOM70依赖性蛋白的导入减少。研究人员通过BN-PAGE进一步探究线粒体膜蛋白复合物的结构完整性,发现在敲减PPID或PERK的细胞线粒体中复合物中TOM70组分减少,而其他组分如TOM40,TIM22和TIM23则未受影响(扩展数据图5b)。PA处理、敲减PERK、敲减PPID均不影响线粒体膜电位(扩展数据图5c),但敲减PPID或联合PA处理均能显著抑制细胞耗氧率(OCR)水平,引起线粒体代谢能量缺陷(图3j)(小编注:研究人员并没有解释为什么OCR水平降低但不影响线粒体膜电位。推测可能的原因是PPID敲减后,抑制TOM70导入,从而抑制ANT2、SLC25A44导入,导致线粒体内ADP、BCAA等底物减少,抑制了电子传递链的底物(NADH/FADH2)生成,从而抑制了OCR水平。但ETC复合体可能仍能通过有限的电子流泵出质子以维持膜电位。或者,由于ANT2缺失,线粒体内ADP减少,ATP合酶活性下降,从而使得质子通过ATP合酶回流减少,能暂时维持膜电位)。总之,这些结果表明,PPID协助TOM70导入OMM,促进线粒体蛋白质向内导入,从而正向调控线粒体呼吸功能。
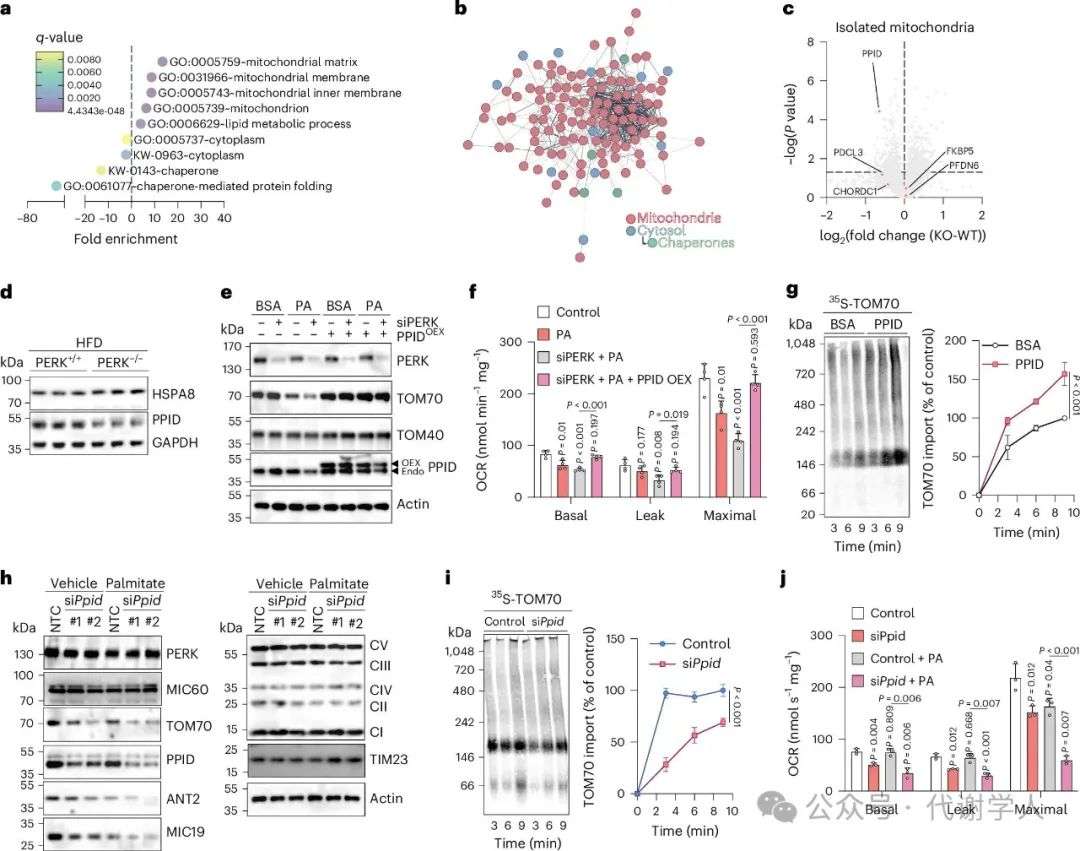
图3.分子伴侣蛋白PPID调节TOM70导入线粒体和呼吸功能
扩展数据图4.高脂饮食压力下,PERK对PPID蛋白调控的作用
扩展数据图5.PPID调控线粒体蛋白质向内运输
3.PPID通过TPR结构域与TOM70相互作用
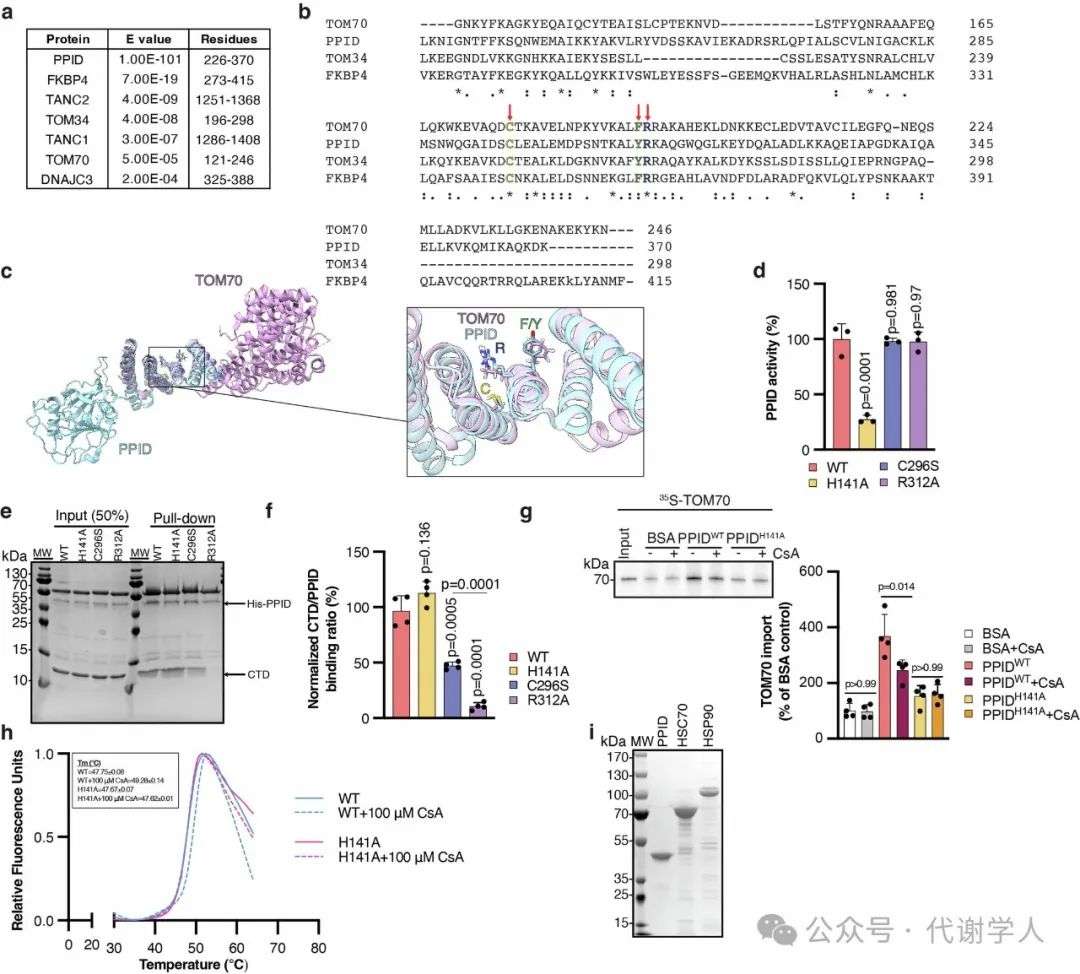
PPID包含两个主要的有序结构域,即N端的肽基-脯氨酰异构酶(PPIase)结构域和C端的TPR结构域,前者负责催化脯氨酸异构化,后者负责识别底物。为了进一步探究PPID和TOM70相互作用的分子机制,研究人员首先探究了PPID是否直接通过物理相互作用来协助TOM70蛋白导入OMM。通过对小鼠蛋白质组中PPID的结构-序列分析,研究人员发现PPID与TOM70以及其他分子伴侣(如FKBPA、TOM34或DNAJC3)的结构域具有相似性(扩展数据图6a,b)。有研究表明,PPID的TPR结构域能与HSP90的C端结构域(CTD)结合,并形成功能性的分子伴侣复合物,进而参与多种蛋白质的折叠和功能调节。而与HSP90相互作用的蛋白中,若含有TPR结构域,则通常存在一段高度保守的序列CX14(F/Y)R。在此,研究人员发现PPID中同样具有高度保守的序列CX14(F/Y)R(扩展数据图6b,c)。接下来,研究人员针对PPID C端的TPR结构域构建C296S、R312A突变体,针对N端的PPIase区域构建H141A突变体(图4a和扩展数据图6d)。为了检测PPIDWT及突变体TPR区域的结合活性,研究人员进行了Pull-down实验,结果显示,R312A突变能完全抑制PPID与HSP90 CTD的结合,但C296S突变仅抑制了50%,表明TPR结构域不同位点的突变会导致不同程度的结合功能失调(扩展数据图6e,f)。随后,研究人员用35S-TOM70和带有重组His标签的PPID进行Pull-down实验,发现只有PPIDC296S突变体能显著与TOM70的结合(图4a)。PPID突变体与HSP90和TOM70的不同结合活性表明TPR识别底物具有特异性。随后,研究人员向分离的线粒体中外源加入PPIDWT和PPIDR312A突变体,均能促进TOM70导入线粒体,但PPIDH141A和PPIDC296S突变体对TOM70的导入没有明显影响(图4b),这说明PPID对TOM70的调控需要N端和C端两个结构域共同作用。同样的,研究人员用脂质体模拟线粒体外膜,发现PPIDWT显著促进了TOM70导入脂质体,而PPIDH141A和PPIDC296S突变体对TOM70的导入没有明显影响,而用环孢素A(cyclosporin A,CsA)抑制PPID PPIase结构域活性后,显著抑制了PPIDWT对TOM70导入的作用(图4c和扩展数据图6g,h)。这进一步证实了PPID PPIase和TPR活性在调控TOM70导入过程中缺一不可。随后,研究人员探究了PPID促进TOM70导入的特异性,发现与HSC70或HSP90相比,PPID能更有效地促进TOM70导入脂质体中(图4d和扩展数据图6i)。因此,PPID能特异性结合TOM70。
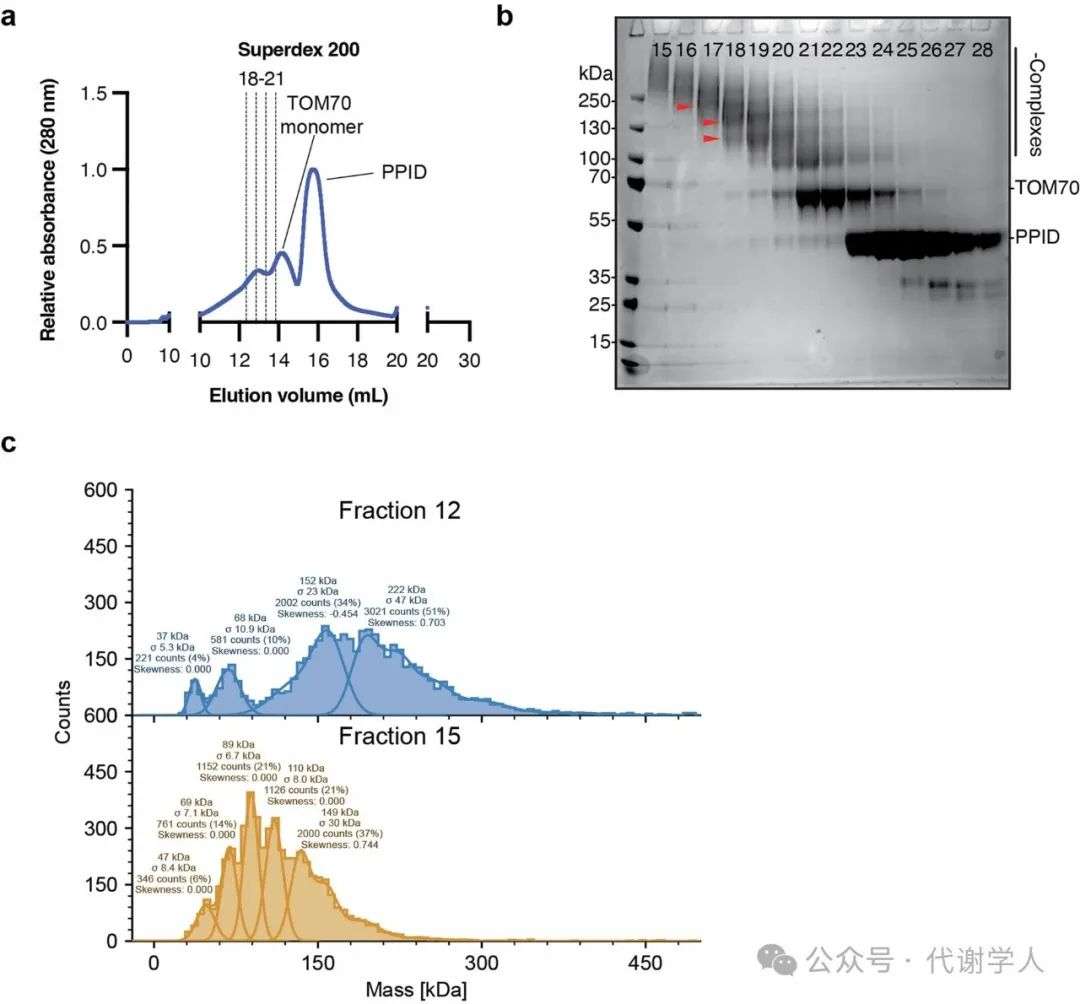
为进一步解析PPID与TOM70的相互作用,研究人员利用重组蛋白在体外组装PPID-TOM70复合物,同时使用SMCC进行温和交联以稳定TOM70二聚体,随后通过尺寸排阻色谱或甘油梯度超速离心对复合物进行分离(图5a和扩展数据图7a,b)。WB结果显示,PPID和TOM70在约100、约150和约200kDa的组分中表现出相似的迁移模式(图5b)。通过光散射质量光度仪(LSMP)检测,发现其质量大小分别为114±8.2、157±28和232±55kDa(扩展数据图7c)。重要的是,与PPIDWT和PPIDR312A相比,PPIDC296S使得PPID-TOM70复合物呈现出明显不同的分布模式,大部分100kDa和150kDa左右的复合物在更早的组分中沉降,且相对含量更高,这说明PPIDC296S突变体使PPID-TOM70复合物的稳定性降低(图5c),进一步证实了PPIDC296S无法有效结合TOM70(图4a-c)。为了解析PPID-TOM70的相互作用方式,研究人员进行了交联质谱(XL-MS)实验。结果表明,PPID C端的TPR结构域与TOM70的C端结构域(CTD)相互作用,而与其N端的分子伴侣钳夹结构域接触较少(图5d)。在棕色脂肪细胞中,研究人员利用Co-IP同样证明了PPID和TOM70的相互作用(图5e)。因此,经生物化学和生物物理学等多种方法证实,多个PPID分子可以同时结合在TOM70的CTD上,提示在TOM70底物结合位点处存在TPR-TPR的稳定机制,同时保留了TOM70膜锚定的N端结构域,使其能够靶向OMM并锚定在膜上(图5d)。总的来说,PPID通过TPR结构域与TOM70特异性结合,从而稳定TOM70,并结合PPIase结构域来促进TOM70导入到OMM上。并且,这种靶向机制与在一种细菌中发现的靶向周质膜的机制类似,因此可能是一种确保膜蛋白正确导入和功能的进化上保守的策略。
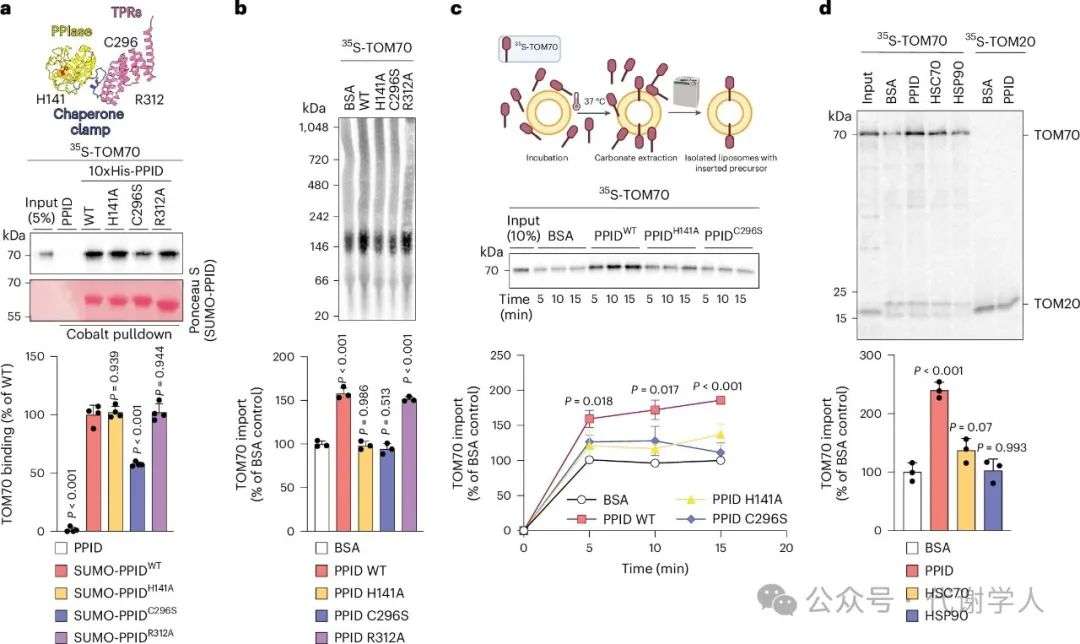
图4.TOM70导入需要PPID的PPIase和TPR结构域
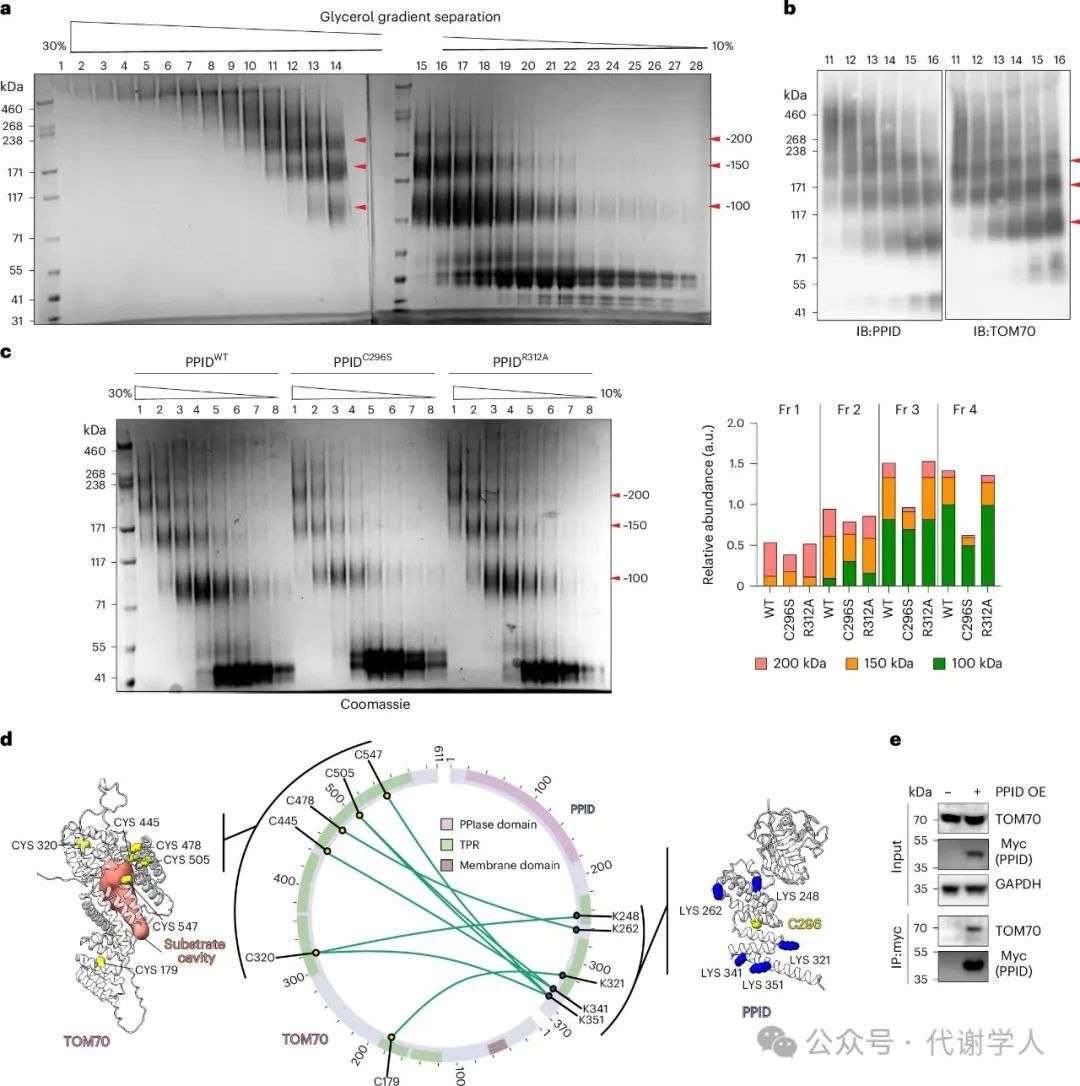
图5.PPID通过其TPR结构域和TOM70相互作用
扩展数据图6.PPID突变体蛋白活性检测
扩展数据图7.PPID-TOM70复合物的体外构建
4.PPID缺失导致小鼠肥胖、血糖异常和线粒体缺陷
上述结果表明,PPID通过直接物理相互作用来调节线粒体功能,介导TOM70导入到OMM。然而,PPID在HFD小鼠中的作用仍未明晰。因此,研究人员构建脂肪细胞特异性PPID-/-小鼠(扩展数据图8a-c)并检测其代谢表型。与HFD PERK-/-小鼠类似,HFD PPID-/-小鼠表现出与糖尿病表型相关的代谢紊乱(图6a-c和扩展数据图8d-i),如体重显著增加(图6a和扩展数据图8d)、体脂增加(扩展数据图8e),葡萄糖不耐受(图6b)、空腹血糖含量增加(图6c)、高胰岛素血症(图6d)和循环游离脂肪酸增加等(扩展数据图8f-h)。在摄食量不变的情况下,HFD PPID-/-小鼠全身耗氧率降低(图6e和扩展数据图8i,j),在热中性条件下对β3-肾上腺素能激动剂CL316243的响应也受到抑制(图6f),以上结果进一步证明了PPID脂肪特异性敲除会导致小鼠的能量和代谢缺陷。同时还发现PPID-/-小鼠的冷耐受明显降低(扩展数据图8k)。这些结果表明,脂肪特异性敲除PPID表现出PERK-/-小鼠类似的表型(图1和扩展数据图1),PPID的脂肪特异性敲除影响HFD小鼠的体温、体重、糖脂稳态。
接下来,研究人员对脂肪特异性敲除PPID小鼠进行组织学分析,发现与PPID脂肪敲除导致体脂增加一致(扩展数据图8e),iBAT和腹股沟白色脂肪组织增大,但肝脏没有增大(图6g和扩展数据图8l)。苏木素伊红染色表明PPID脂肪敲除后iBAT组织变白,脂滴变大(图6h),电镜结果也提示线粒体嵴密度降低(图7a和扩展数据图9)。WB检测发现PPID敲除导致iBAT中TOM70及其底物ANT2、SLC25A44、UCP1和MIC19等蛋白水平下调,说明蛋白质稳态受损(图7b)。此外,与此前结果一致,35S-TOM70 在PPID-/-小鼠iBAT的线粒体中的导入减少(图7c),线粒体呼吸功能也显著受损,即使用FCCP处理也无法增加耗氧率(图7d,e)。总之,PPID能正向调节线粒体呼吸和代谢功能,可以促进TOM70在OMM上的导入,促进线粒体相关蛋白向内转运。在饮食诱导的肥胖期间,PERK依赖性的PPID伴侣蛋白活性降低会导致线粒体蛋白质无法向内转运,呼吸和产热功能失衡,进而影响体温、体重和葡萄糖稳态。
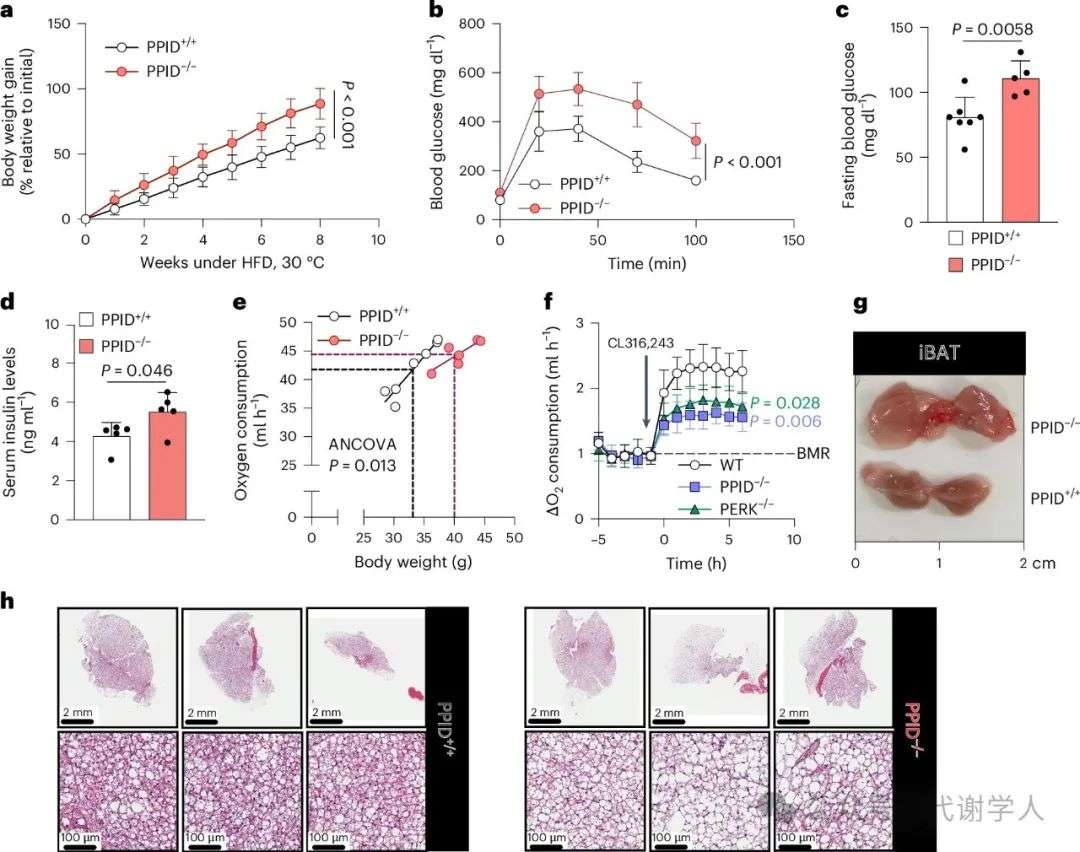
图6.PPID调控HFD小鼠体内TOM70的膜导入和线粒体功能
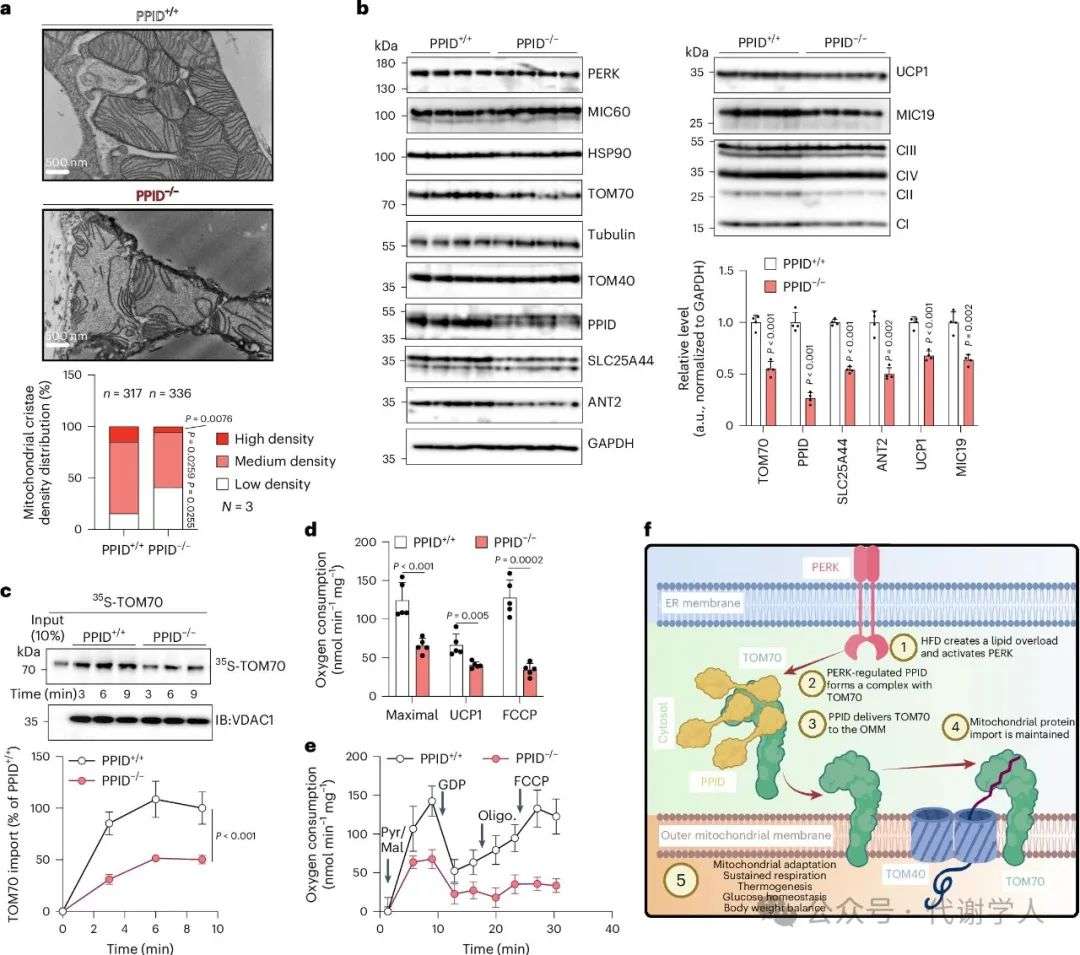
图7.PPID调控HFD小鼠体内TOM70的膜导入和线粒体功能
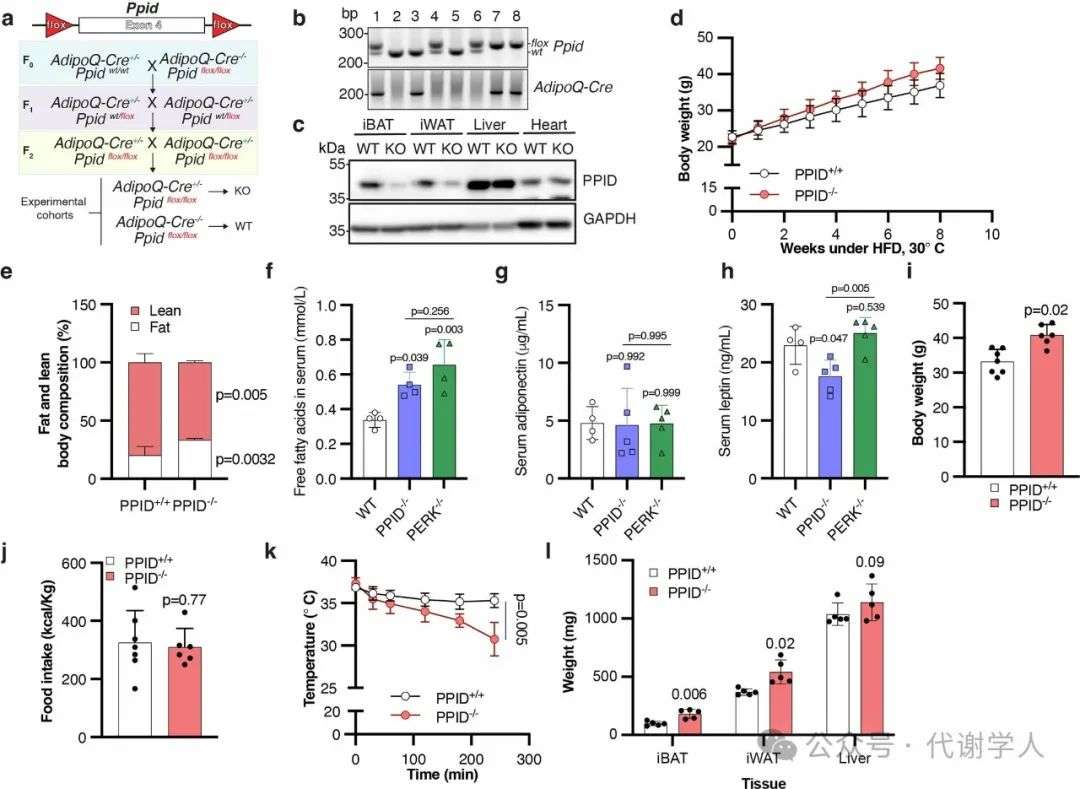
扩展数据图8.PPID敲除对HFD小鼠脂肪细胞的影响
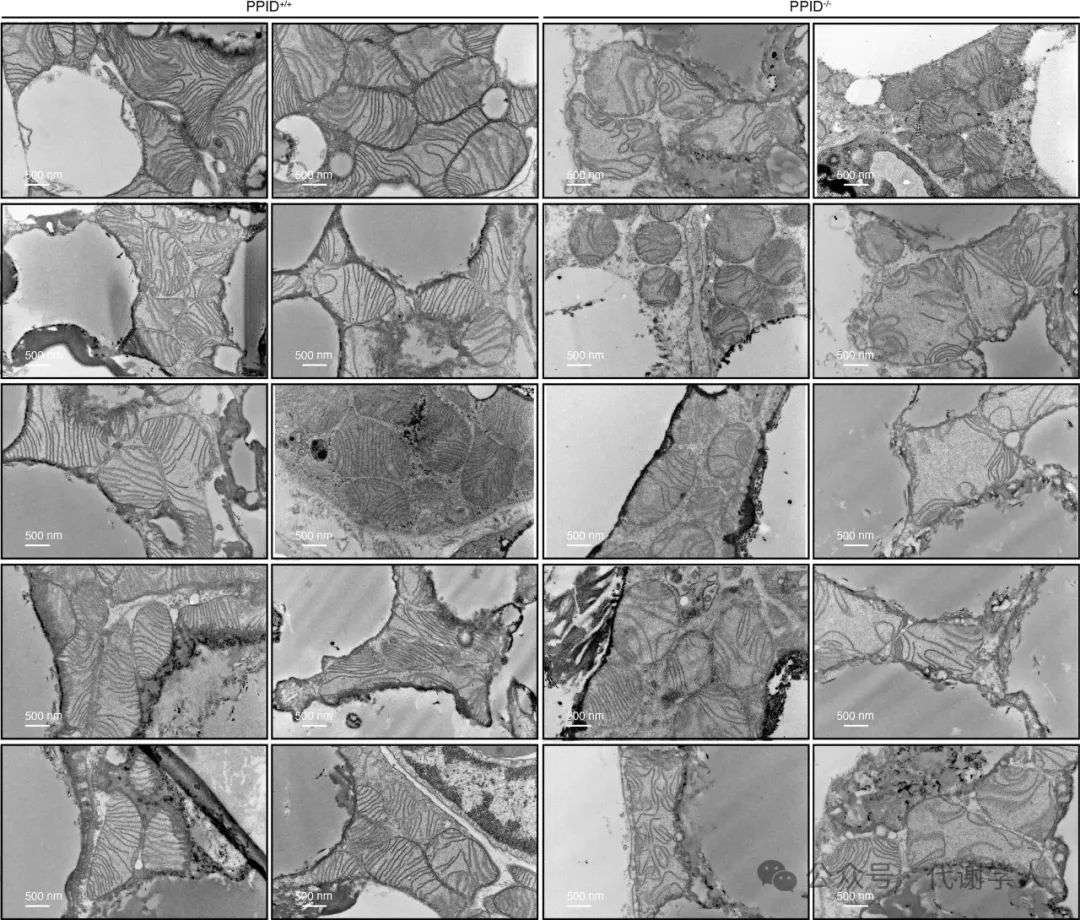
扩展数据图9.电镜下HFD PPID-/-小鼠iBAT中线粒体的超微结构
总结:
本文中,研究人员发现PERK依赖性细胞质分子伴侣PPID能响应内质网应激,驱动TOM70导入OMM,促进线粒体相关蛋白向内转运,进而调控棕色脂肪细胞的线粒体呼吸和产热功能。进一步研究发现,PPID通过TPR结构域与TOM70特异性结合,从而稳定TOM70,并结合PPIase结构域来促进TOM70导入到OMM上,进而调控线粒体功能。总之,本篇文章在OMM蛋白导入的分子机制方面提供了新的见解,揭示了PPID在维持线粒体功能和代谢健康中的关键作用,并为治疗肥胖和2型糖尿病提供了新的潜在靶点。
链接:https://www.nature.com/articles/s41556-024-01555-z
https://wap.sciencenet.cn/blog-3483272-1478814.html
下一篇:Science:糖尿病,“细胞身份丢失”引起的全身崩溃