博文
抑制线粒体融合以防止线粒体DNA逃逸  精选
精选
|
抑制线粒体融合以防止线粒体DNA逃逸
线粒体起源于一种细菌。真核细胞是两种细菌融合的后代,其中线粒体是细菌,真核的其他部分主要来自一种古菌。作为细菌的后代,线粒体依靠自身分裂产生后代,但也具有一种细菌不具备的生存方式,就是能够相互融合。在融合过程中,可产生一些非常肥大的线粒体,而这种线粒体能持续释放出线粒体DNA。这种来自线粒体的DNA,可以被细胞作为外来物质,类似于普通细菌感染的危险信号分子,启动炎症反应。严重情况下可以导致细胞死亡甚至疾病发生。这里就有能提出一种疾病治疗新策略,通过阻断线粒体融合,控制线粒体DNA释放,实现治疗疾病目的。
被称为线粒体的细胞器含有DNA,并且能够相互融合或分裂。事实表明,应激激活的调节因子可以避免过度融合,从而防止线粒体DNA潜在的有害释放。
我们细胞的“动力工厂”是含有DNA的细胞器,即线粒体。这些细胞器存在于一个网络中,在线粒体网络里,线粒体可以分裂或相互连接。山田等人在《自然》杂志上发表的文章揭示了一个通过调节线粒体融合来帮助防止线粒体DNA释放到细胞质中的系统。
线粒体是由一种被内化的细菌进化而来的,祖先线粒体基因组中的大多数基因已经转移到了细胞的细胞核中。人类线粒体DNA(mtDNA)仅编码13种蛋白质,其余的(大约99%)由核基因编码并被导入线粒体中。这些细胞器通常与细胞的其他部分协同工作,受到强大的线粒体外膜和内膜的保护。但是,当这些膜受损时,线粒体成分会释放到细胞质中,给细胞带来可怕的后果。这是因为细胞中识别细菌感染的通路也会被线粒体物质激活,从而引发免疫反应或迅速诱导细胞死亡。
存在多种途径来控制线粒体。例如,当线粒体内膜上的电荷(膜电位)被破坏时,线粒体酶OMA1会被激活,并切割信号蛋白DELE1。被切割的DELE1进入细胞质并刺激细胞的整合应激反应,这种反应要么有助于重新调整细胞的代谢以促进恢复,要么在情况无法恢复时促使细胞走向死亡。
然而,蛋白质PINK1和Parkin会同时启动一个更具选择性的维护途径。这个维护途径通过一种称为自噬的降解过程来消化单个受损的线粒体。Parkin是一种称为E3泛素连接酶的酶,它存在于细胞质中,并且和OMA1一样,在受到线粒体应激时会被激活。线粒体膜电位的丧失会导致PINK1(一种激酶类酶)在线粒体外膜上积累。这会促使Parkin用泛素分子覆盖线粒体表面,为受损的细胞器贴上标签以便进行自噬。值得注意的是,人类Parkin或PINK1的突变是一种早发性帕金森病最常见的病因。
尽管OMA1、PINK1和Parkin似乎是控制线粒体过程中的关键角色,但经过基因工程改造而缺失编码这些蛋白质(OMA1、PINK1或Prkn)的相应基因之一的小鼠,总体上是正常的。这表明OMA1或PINK1与Parkin的作用可能仅在应激条件下才至关重要,或者其他途径可以弥补它们各自缺失所带来的影响。
为了进一步研究,山田等人培育出了缺失OMA1且缺失Parkin或PINK1的小鼠。这些小鼠比正常小鼠体型更小,运动功能下降,身体颤抖,并且与保留这些基因的动物相比,寿命显著缩短。从表面上看,OMA1和Parkin介导的途径在很大程度上没有关联,仅通过它们对线粒体应激的反应性联系在一起。单个基因缺失本身没有重大影响,但两个基因同时缺失却会产生如此有害的后果,这一事实表明这两条途径之间存在协同作用,并且会汇聚到相同的反应上。但这种反应是什么呢?
缺失Parkin和DELE1的小鼠并没有表现出缺失Parkin和OMA1的小鼠的特征,这表明与Parkin协同作用的途径不是整合应激反应。这促使作者研究OMA1的另一个潜在靶点,内膜蛋白OPA1,它是线粒体融合的关键调节因子。
细胞中呈细长分支状网络的线粒体不断经历分裂和融合过程。平衡这些相反的过程对于细胞健康至关重要,影响线粒体动态变化的突变会导致疾病。线粒体分裂产生新的细胞器,使受损线粒体能够分离和循环利用(包括通过自噬),并且在细胞应激时可以促进细胞死亡。相反,线粒体融合允许蛋白质和线粒体DNA的混合与交换,以维持整个网络的一致性。激活的OMA1会切割具有融合活性的长链形式的OPA1,产生无融合活性的短链形式,从而使平衡向分裂方向倾斜。
线粒体融合主要由三种称为GTP酶的酶控制:OPA1以及外膜蛋白MFN1和MFN2。相邻线粒体上的MFN1和MFN2之间的相互作用使两个细胞器靠近,促使外膜融合,然后OPA1协调内膜的融合。
为了探究缺失Parkin和OMA1的小鼠出现的严重症状是否是由于线粒体融合的变化导致的,山田及其同事通过删除Opa1或Mfn1基因的两个拷贝中的一个(完全去除Opa1或Mfn1是致命的)来降低融合活性。令人惊讶的是,这恢复了这些动物的体重和生存缺陷,并且这些小鼠没有出现运动问题,这表明这些特征不是由抑制的应激反应驱动的,而是由过度的线粒体融合导致的。
通过使用各种显微镜观察方法,山田等人在缺失Parkin和OMA1的小鼠的大脑和心脏中观察到了一小部分增大的线粒体,称为巨型线粒体(图1)。这些增大的细胞器的内膜褶皱(称为嵴)比正常线粒体少。有趣的是,尽管存在这些巨型线粒体,线粒体群体在很大程度上仍保持代谢功能,这就提出了一个问题,即线粒体网络的这些变化如何会导致如此明显的生长缺陷。
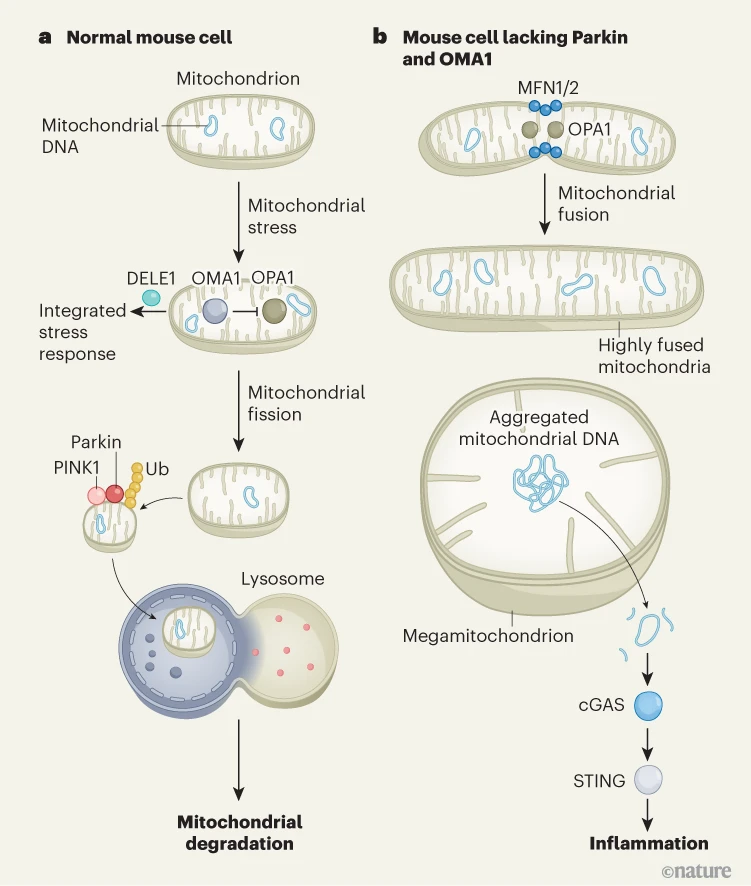
图1 | 过度的线粒体融合引发细胞炎症。含有DNA的线粒体细胞器通常存在于一个动态网络中,在这个网络中,这些细胞器会经历分裂和融合事件。a,在线粒体应激期间,蛋白质OMA1会将通常促进融合的蛋白质OPA1切割成无融合活性的形式,从而抑制OPA1并使线粒体网络的平衡向线粒体分裂方向倾斜。与此同时,Parkin蛋白会被PINK1蛋白招募到线粒体外膜上。Parkin将泛素(Ub)蛋白添加到细胞器上,为其贴上标签以便通过称为自噬的过程进行降解,在自噬过程中,线粒体物质与一种称为溶酶体的细胞器融合。另一个对线粒体应激做出反应的途径涉及蛋白质DELE1,它会激活整合应激反应。b,山田等人描述了一些实验,在这些实验中,经过基因工程改造而缺失Parkin和OMA1的小鼠使线粒体动态变化的平衡向由MFN1、MFN2和OPA1蛋白介导的过度融合方向倾斜。这些小鼠某些组织中的细胞含有一小部分极大的线粒体(“巨型线粒体”),其中含有线粒体DNA聚集体。通过一种尚未确定的机制,含有巨型线粒体的细胞将线粒体DNA释放到细胞质中。这会通过蛋白质cGAS和STING激活炎症免疫反应。
作者在缺失Parkin和OMA1的小鼠大脑中发现了一种与免疫反应相关的基因表达特征。降低OPA1或MFN1的水平可以防止这种特征出现,并且没有观察到巨型线粒体。鉴于许多线粒体成分,包括线粒体DNA,保留了与其细菌起源相关的特征,因此在细胞器外是强大的免疫触发因素,山田等人将研究重点放在了线粒体DNA的位置上。
果然,在巨型线粒体中发现了异常的线粒体DNA聚集体,并且一些线粒体DNA逃逸到了细胞质中。在细胞质中,一种称为cGAS的酶可以识别这种线粒体DNA,cGAS也能识别细菌DNA。cGAS会激活STING蛋白,从而启动细胞免疫反应。令人惊讶的是,在缺失Parkin和OMA1的小鼠中,通过基因工程手段使它们额外缺失STING,克服了这些动物的生长缺陷和寿命短的问题。这些发现共同表明,Parkin和OMA1可以防止过度的线粒体融合和线粒体DNA的释放,从而防止引发严重降低小鼠生长、运动功能和寿命的免疫反应。
这并不是首次将异常的线粒体动态变化与线粒体DNA释放联系起来,但关键的是,现在已经直接将其与体内的严重症状联系起来了。先前的体外研究报告称,失调的分裂(不足或过度)、紊乱的嵴结构或YME1L(另一种可以加工OPA1的酶)的缺失,都是可以通过多种机制诱导线粒体DNA释放的事件。
过度的线粒体融合是如何导致线粒体DNA释放的呢?线粒体DNA是从已形成的巨型线粒体中释放出来的,还是从正在形成或降解的线粒体中释放出来的呢?已经描述了多种线粒体DNA释放的机制,但尚不清楚在缺失Parkin和OMA1的小鼠中,这些机制(如果有的话)哪些会起作用。此外,巨型线粒体的存在和随之而来的炎症反应是具有组织特异性的,这就提出了一个问题,即在未受影响的组织中,哪些途径会阻止过度融合。存在多种不依赖于PINK1/Parkin的线粒体自噬机制。这些机制中的任何一种能否保护未受影响的组织呢?
这些发现为越来越多的研究增添了新的内容,这些研究表明线粒体应激和STING介导的神经炎症可能在神经退行性疾病中发挥作用。例如,Parkin的缺失,再加上线粒体DNA酶POLG的突变,会在小鼠身上引发严重的类似帕金森病的症状。如果这些动物也缺乏STING,这种症状就会得到预防。在阿尔茨海默病的小鼠模型中,β淀粉样蛋白和tau蛋白会诱导线粒体应激和线粒体DNA释放,抑制STING可以减轻这些小鼠的许多疾病症状。神经退行性疾病的复杂性意味着大多数小鼠模型无法重现所有的临床症状,缺失Parkin和OMA1的小鼠也不例外。然而,这些模型确实提供了诱人的证据,表明线粒体动态变化对神经退行性疾病的影响值得进一步研究。
https://wap.sciencenet.cn/blog-41174-1474087.html
上一篇:饮用氢气纳米气泡水辐射防护作用研究【日本2025】
下一篇:静电的奥秘终于得以揭示