博文
控制程序性坏死治疗病毒感染后肺损伤和全身炎症  精选
精选
||
要从大流行病中学到的一个教训是,我们需要为下一次做好更充分的准备。流感大流行在过去已经导致数百万人死亡,而且可能再次发生。与冠状病毒SARS-CoV-2相比,一些流感毒株对年轻、表面上健康的成年人特别致命,这可能是因为在这些人群中引发了强烈的炎症反应。
Gautam等人在《自然》杂志上发表的文章中表明,通过抑制一种称为坏死性凋亡的细胞死亡反应——一种重要的先天抗病毒策略——在严重流感感染的动物模型中可以减少炎症反应的强度,从而大幅降低死亡率。
尽管先天免疫系统分支释放的激烈炎症反应显然会限制病毒生长,但在肺部感染中,反应的强度和关键炎症分子(称为细胞因子)的水平实际上是重症疾病最明确的预测指标之一。直观上,人们可能会认为这是因为炎症反应的强度反映了病毒量(病毒载量)。然而,来自呼吸道组织的样本中的病毒载量并不是疾病严重程度的可靠预测指标。
强烈炎症反应的预测能力可能源于这样一个事实:除了帮助对抗感染外,炎症反应还可以干扰受感染组织的功能。这表明需要在这两个结果之间保持微妙的平衡。过度和破坏性的炎症反应,或称为“细胞因子风暴”,在COVID-19的背景下已经被讨论过。当前的研究不仅表明坏死性凋亡可以驱动炎症,而且还提供了与假设一致的证据,即细胞因子风暴是重症疾病的原因,而不仅仅是相关因素。
病毒需要活体宿主细胞才能复制,因此细胞自杀作为对感染的反应是一种特别有效的抗病毒策略。一种称为凋亡的机制,由称为半胱天冬酶的酶介导,是首选的细胞死亡反应。这可能是因为经历凋亡的垂死细胞被其他细胞迅速吸收,防止它们的细胞内容物释放到周围组织中,从而实现对炎症反应的严格控制。病毒已经进化出阻止凋亡的策略,这在进化的军备竞赛中,迫使宿主做出相应的反应。其中一种策略是坏死性凋亡——一种垂死细胞破裂的细胞死亡类型——通常在半胱天冬酶被抑制时激活。然而,与通过凋亡导致的细胞死亡相比,坏死性凋亡似乎伴随着引起更多组织损伤的炎症反应风险。
坏死性凋亡是由与引发凋亡相似的刺激触发的。这些包括病毒感染的迹象,例如病毒RNA的存在。在流感感染中,宿主蛋白ZBP1结合到一种称为Z-RNA的病毒RNA,并激活RIPK3,这是一种可以将磷酸基团加到蛋白质上的酶,称为激酶,这个过程被称为磷酸化。然后,RIPK3以非依赖激酶的方式,通过蛋白质半胱天冬酶-8激活凋亡途径。在流感感染期间,与正常的坏死性凋亡激活顺序过程不同,该过程只有在凋亡被抑制时才会发生,激活的RIPK3磷酸化并激活坏死性凋亡所需的蛋白质,即形成孔的分子MLKL(图1)。
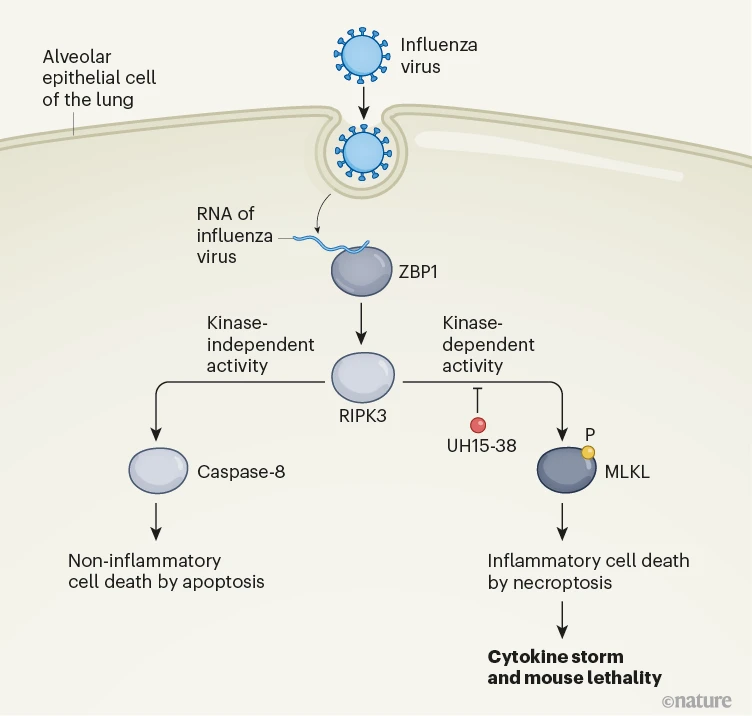
图1 | 预防与流感感染相关的肺损伤的抑制剂分子。覆盖肺部表面的肺泡上皮细胞可以在防御反应中死亡,以防止病毒传播。流感病毒的RNA被蛋白质ZBP1识别。这可以激活蛋白质RIPK3,它是一种激酶,可以将磷酸(P)基团加到目标蛋白质如MLKL上。RIPK3可以以非依赖激酶的方式激活蛋白质半胱天冬酶-8,触发一种称为凋亡的非炎症性细胞死亡。RIPK3还可以以依赖激酶的方式启动一种更具破坏性的细胞死亡,称为坏死性凋亡。这与破坏性炎症和释放的炎症分子(未显示)相关,产生所谓的细胞因子风暴。Gautam等人报告说,一种名为UH15-38的RIPK3激酶活性抑制剂可以阻断与流感感染相关的坏死性凋亡,减少组织损伤,并在小鼠中预防致死性。
尽管这种细胞死亡途径的双重激活,凋亡途径足以限制病毒生长并支持正常的免疫应答3,这使得通过抑制RIPK3的激酶活性来阻断坏死性凋亡成为流感感染期间一个有吸引力的治疗靶点。不幸的是,无论是通过小分子还是基因手段抑制RIPK3激酶活性,通常都会激活凋亡4,5,这降低了对这种策略的热情。Gautam等人报道了一种名为UH15-38的强效RIPK3抑制剂的开发,它在低剂量下能抑制坏死性凋亡但不激活凋亡。此外,该抑制剂对其他炎症信号通路没有任何非靶向活性,也不抑制密切相关的激酶。
每天注射UH15-38的小鼠耐受良好,没有明显的毒性迹象,关键是该抑制剂在降低流感感染模型中的死亡率方面非常有效。此外,UH15-38并没有提高缺乏RIPK3或MLKL的感染小鼠的存活率,这表明抑制剂的效果是由于靶向活性所致。与另一种实际上增加了疾病严重程度的RIPK3抑制剂相比,UH15-38也更有效地保护了小鼠,这可能是由于抑制剂具有在未感染细胞中诱导凋亡的次要能力。
作者报告的一个有趣发现是,在感染开始后,一种称为I型肺泡上皮细胞的特殊类型的肺细胞含有比其他任何类型肺细胞多大约80倍的病毒RNA。这与之前观察到的特定细胞类型的破坏——从这些细胞的10%破坏阈值开始——与肺功能丧失和致死性相关联6相一致。与UH15-38的效果和使用UH15-38进行疾病治疗的潜在重要性一致,这些细胞表达了所有所需的坏死性凋亡机制,并且在感染时,MLKL发生磷酸化,这一过程可以被UH15-38阻断。相比之下,半胱天冬酶-8和半胱天冬酶-3的激活不受UH15-38的影响。
作者提出的最引人注目的发现是UH15-38在感染后至少5天内有效。在早期的小鼠研究中,临床上批准使用的抗病毒药物,如奥司他韦和扎那米韦,最好在感染前(预防性)给药,如果在感染开始后48小时给药则不提供显著保护7,8。因此,这些药物通常只推荐给在症状首次出现后48小时内的高风险患者。如果这两种类型的抑制剂能够直接对比测试,以确定UH15-38的优越性是否可以得到确认,以及这些发现是否与临床治疗相关,那将是很有趣的。
UH15-38在流感中特别有效是因为它积聚在肺部还是因为肺部特别容易发生坏死性凋亡?两者都有可能。Gautam及其同事报告说,UH15-38在肺部的水平比血浆中的水平高八倍。已有多份报告关于其他肺部疾病,包括慢性阻塞性肺病和哮喘,至少在小鼠模型中,已显示坏死性凋亡有助于疾病的严重程度9。相反,一篇研究SARS-CoV-2感染中坏死性凋亡的作用的论文,基于对缺乏MLKL的小鼠致命感染的研究,表明坏死性凋亡与疾病严重程度无关10,这表明抑制坏死性凋亡不会是所有呼吸性疾病的万能药。
因此,使用这种新的RIPK3抑制剂来应对流感感染取得了平衡,减轻了炎症反应的力量但保持了其抗病毒效果。我们急切地期待着可能有助于下一次大流行的临床试验。
严重流感A病毒(IAV)感染可能导致过度炎症、肺部损伤和急性呼吸窘迫综合征1,2,3,4,5 (ARDS),目前没有有效的药物治疗方法。程序性坏死是治疗ARDS及相关炎症疾病的一个有吸引力的切入点,因为它在严重的IAV感染期间推动病理性的肺部炎症和致死性6,7,8,并且可以通过受体相互作用蛋白激酶3 (RIPK3)抑制剂来潜在地针对它。在这里,我们展示了一个新开发的RIPK3抑制剂UH15-38,在体内强有力且选择性地阻断了IAV触发的肺泡上皮细胞中的程序性坏死。UH15-38改善了肺部炎症,并在感染实验室适应性和大流行株系的IAV后预防了死亡,而不影响抗病毒的适应性免疫反应或妨碍病毒清除。即使在感染过程中晚期给药,UH15-38也显示出强大的治疗效果,这表明RIPK3阻断可能在患有IAV驱动的ARDS和其他过度炎症病理学的患者中提供临床益处。
主体
季节性IAV感染每年导致全球多达500万例严重疾病,造成多达65万人死亡。尽管迄今为止在人类之间传播的高致病性H3、H5和H7禽流感A病毒株有限,但它们可能只需要少量突变就能变得可传播并实现大流行潜力9,10,11。由于目前的疫苗和抗病毒策略要么效果有限,要么容易受到病毒抗性和逃避的影响,因此迫切需要确定新的治疗恶性IAV引发的肺部疾病的方法。
IAV激活的坏死性细胞死亡可能是这样一个切入点。作为一种裂解性病毒,IAV杀死了大部分它在其中复制的肺细胞类型12。当这种死亡得到良好控制时,它是病毒清除的有效机制,但当死亡不受检查或主要是坏死性的时候,尽管病毒被清除,肺部损伤和严重疾病仍会发生12,13。在小鼠模型中观察到这种严重的病理学,其中气道上皮的破坏是致命IAV感染的标志4,14,15,在人类中也是如此,其中远端肺上皮的广泛死亡,以支气管肺泡坏死区域为特征,是IAV诱导的ARDS以及病毒性和继发性细菌性肺炎的典型特征2,3,12。
程序性坏死推动了流感的严重程度
程序性坏死占据了大多数由IAV激活的感染肺上皮细胞中的程序性坏死性死亡6,7,8,12,16,17。程序性坏死是由宿主传感器蛋白Z-形核酸结合蛋白1 (ZBP1)启动的,它检测到IAV产生的Z-RNA并激活RIPK318,19。然后RIPK3磷酸化混合谱系激酶样蛋白(MLKL),从而诱导程序性坏死6,8,12,13,18 (图1a)。程序性坏死触发了严重的IAV感染期间的肺组织坏死、病原性中性粒细胞招募和肺部炎症,但对于CD8+ T细胞介导的抗病毒反应或病毒清除来说并不是必须的8,13。这是因为RIPK3还激活了一个平行的凋亡途径,即使在没有程序性坏死信号的情况下也完全有能力限制IAV6。这些观察将程序性坏死定位为治疗干预的一个有吸引力的节点,因为只有程序性坏死而不是凋亡依赖于RIPK3激酶功能6,20 (图1a)。预计RIPK3激酶活性的抑制剂可以改善坏死性肺损伤而不影响病毒清除,因此可能代表治疗IAV引发的肺部炎症和损伤的策略。
网络上各种死亡形式名词。
1、坏死(Necrosis):一种非程序化的、不受调节的细胞死亡过程,其特征是细胞肿胀、生物膜完整性丧失、细胞内容物溢出和离子梯度的消散,从而引发炎症反应。
2、细胞凋亡(Apoptosis)、坏死性凋亡(Necroptosis)、细胞焦亡(Pyroptosis)与铁死亡(Ferroptosis),详见“细胞死亡方式与肿瘤研究热点的碰撞”。
3、铜死亡(Cuproptosis):详见“Science|Todd R Golub团队带你了解铜死亡”。
4、细胞胀亡(Oncosis):1910年,vonReckling-hausen首次发现一种因骨软化症而肿胀、坏死的骨细胞,1995年,Majno和Joris提出了Oncosis的概念,将具有明显肿胀特征的细胞死亡称为Oncosis。胀亡细胞周围有明显炎症反应。
5、自噬(Autophagy):百度百科的解释为,由Ashford和Porter在1962年发现细胞内有“自己吃自己”的现象后提出,是指从粗面内质网的无核糖体附着区脱落的双层膜包裹部分胞质和细胞内需降解的细胞器、蛋白质等成分形成自噬体(autophagosome),并与溶酶体融合形成自噬溶酶体,降解其所包裹的内容物,以实现细胞本身的代谢需要和某些细胞器的更新。
6、失巢凋亡(Anoikis):是1994年首次命名的一种特殊的程序化细胞死亡形式,由细胞与细胞外基质(Extracellular matrix,ECM)和其他细胞失去接触而诱发,caspase作为关键信号通过依赖线粒体和caspase活化影响细胞死亡,是失巢凋亡的重要机制。
7、副凋亡(Paraptosis):2000年Sperandio等在293T细胞系中超表达胰岛素样生长因子1受体(IGFIR)时发现的一种非凋亡性细胞程序死亡,细胞浆空泡化,线粒体和内质网肿胀,但没有核固缩现象。
8、免疫原性细胞死亡(Immunogenic cell death,ICD):2005年提出的用于描述可以引起免疫应答的细胞死亡。ICD是调节性细胞死亡的一个特定变体,由应激压力驱动,可诱导针对死亡细胞抗原的适应性免疫。
9、细胞套亡(Entosis):是2007年首次报道的一种细胞死亡新形式,将细胞能够吞噬整个活细胞和/或濒死细胞的这种非吞噬过程称为“细胞套亡”,有可能成为一种抑制肿瘤的新方法。
10、Parthanatos:又称PARP-1依赖性细胞死亡,2007年由约翰霍普金斯大学医学院Ted Dawson与Valina Dawson夫妻发现的一种基于DNA损伤,PARP-1激活的程序性细胞坏死形式,是一个受到调控过程,由PAR聚集、线粒体凋亡诱导因子AIF转移到细胞核内引起的,它会导致大片段的DNA碎片产生,且不依赖caspase。
11、泛凋亡(PANoptosis):最早在2016年被报道,基于对炎症小体/焦亡与凋亡和坏死性凋亡之间的交互作用的研究而建立,是一种炎症性程序细胞死亡,受PANoptosome(泛凋亡体)复合物调控,具有Pyroptosis、Apoptosis和/或Necroptosis的关键特征,这也是PANoptosis术语中“P”、“A”和“N”的来源,不可被细胞焦亡、凋亡和坏死性凋亡中任意一种死亡方式单独表征。
12、双硫死亡(Disulfidptosis):2023年,德克萨斯大学MD安德森癌症中心甘波谊教授和陈俊杰教授团队发现的一种新的细胞死亡机制,由细胞内过量胱氨酸积累引起的二硫化物应激(disulfide stress)导致的快速死亡方式,通常在葡萄糖饥饿的条件下发生。
https://wap.sciencenet.cn/blog-41174-1429255.html
上一篇:释氢珊瑚钙对甲基苯丙胺戒断表现的作用
下一篇:发现真核细胞固氮能力的细胞器硝质体