博文
[转载]hLife | Ashif Iqubal等研究团队阐明小胶质细胞与阿尔茨海默病的神经炎症和认知障碍的关系
||
阿尔茨海默病(AD)是导致老年人痴呆的主要原因,约有近10%的65岁以上人群和30%的85岁以上人群患病。研究表明,多种病理机制和信号分子可能参与疾病的发生与发展,其中神经炎症被认为是AD的重要标志之一。淀粉样蛋白β(Aβ)、过度磷酸化的tau蛋白及其形成的神经纤维缠结(NFT)与神经炎症密切相关。小胶质细胞的激活被证实是推动神经炎症及疾病进展的关键因素。
近日,印度Jamia Hamdard大学Ashif Iqubal等研究团队在hLife发表了题为“Mechanism of microglia-mediated neuroinflammation, associated cognitive dysfunction, and therapeutic updates in Alzheimer's disease” 的文章(图1),详细梳理了小胶质细胞激活机制及其与神经炎症和痴呆的关系,并介绍了作用于tau蛋白或缓解神经炎症的药物及其临床进展。

图1 论文标题及作者信息
聚焦神经炎症
近年来,神经炎症在AD发病机制中的作用受到越来越多关注,并被认为可能是由Aβ和tau蛋白的积累所触发。胶质细胞(包括星形胶质细胞和小胶质细胞)是这一炎症反应的核心调控者,相关炎症标志物可在疾病早期被检测到,因而也成为新的治疗靶点。AD的病因复杂,还涉及氧化应激、线粒体功能障碍和细胞凋亡等多个方面,调控小胶质细胞活化被认为是开发新型抗AD药物的重要策略之一。
小胶质细胞在AD中的角色
小胶质细胞是大脑中的常驻巨噬细胞,构成神经免疫系统的第一道防线,在正常情况下具有神经保护作用,参与维持稳态、调节代谢动态以及支持突触可塑性、学习与记忆功能(图2)。然而,在受到神经毒性刺激、遗传因素或病原体影响时,小胶质细胞会被激活,进而引发神经炎症,与AD、抑郁、痴呆及认知障碍等疾病密切相关。老化是小胶质细胞活化的重要促进因素,老年人的小胶质细胞更容易被激活,释放更多促炎因子(如IL-6和TNF-α),且清除Aβ的能力下降,对INF-γ等刺激的反应也更为敏感,倾向于进入促炎状态。图2展示了小胶质细胞的两种主要功能表型:具有吞噬功能和支持神经健康的M2型(神经保护型),以及释放有害细胞因子、破坏突触功能的M1型(促炎神经毒型)。从M2向M1表型的转变是神经退行性病变的重要机制,因此,针对小胶质细胞的AD治疗策略主要聚焦于抑制其整体活化或阻止其向M1型极化,从而实现神经保护作用。
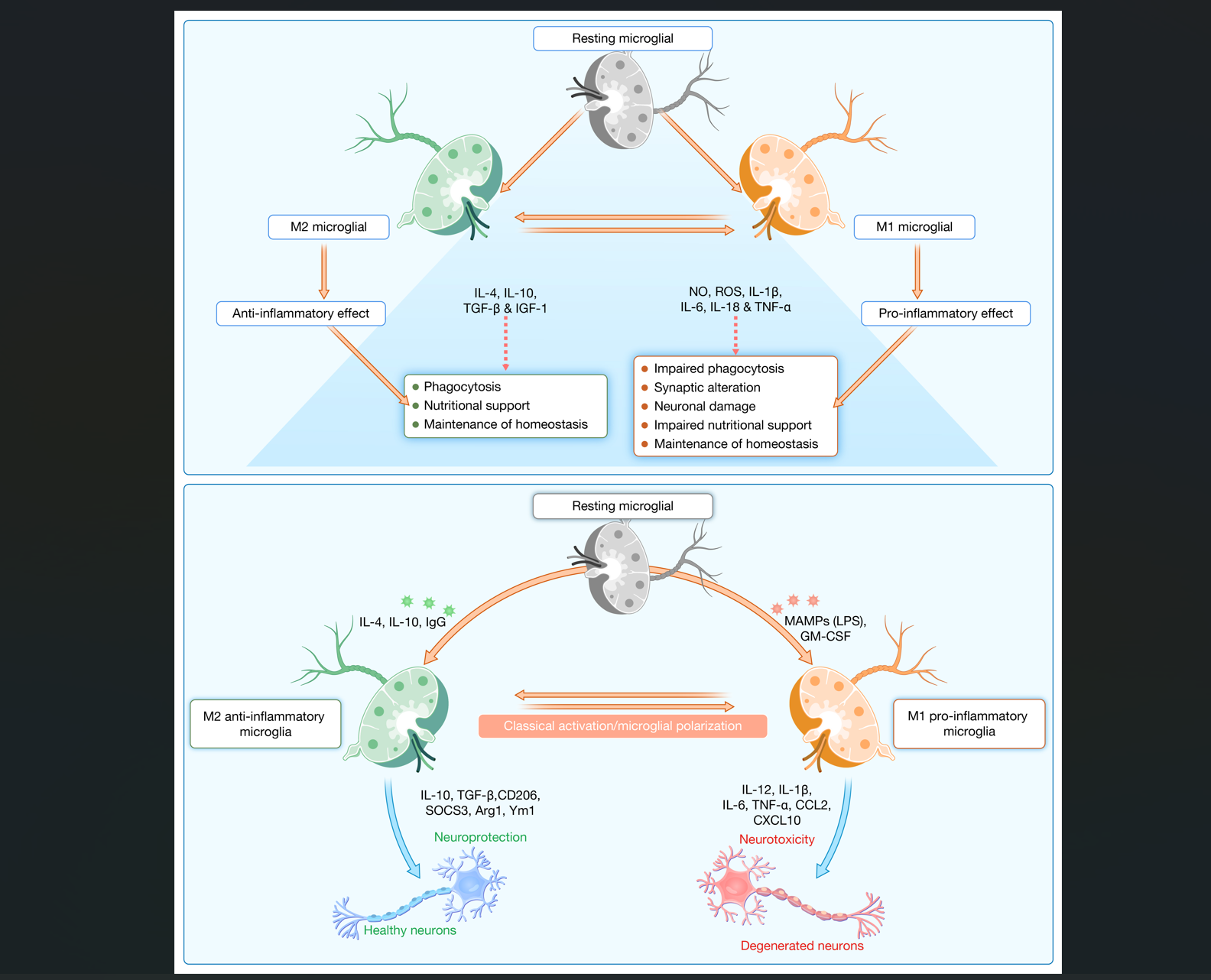
图2 小胶质细胞在AD的生理学与病理学过程中的作用
小胶质细胞在AD中被激活
小胶质细胞作为中枢神经系统的主要先天免疫细胞,调节大脑炎症反应。其M1表型通过诱导核因子-κB(NF-κB)和诱导型一氧化氮合酶(iNOS)途径,产生如TNF-α、ILs等促炎细胞因子以及活性氧(ROS),M2表型则执行吞噬作用和支持神经元健康。在AD中,小胶质细胞的过度激活导致神经炎症,伴随有Aβ和神经纤维缠结(NFT)的形成与沉积,这不仅对神经元造成损伤,还引起认知功能下降。此外,当小胶质细胞无法分解tau蛋白时,含有tau的外泌体会释放并穿透神经元,影响线粒体活动,加剧神经退行性变和突触损伤。
激活小胶质细胞的刺激因素
多种刺激物可以激活小胶质细胞,包括Toll样受体-4(TLR-4)、模式识别受体(PRRs)、病原体相关分子模式(PAMPs)或损伤相关分子模式(DAMPs)等。这些分子能够感应PAMPs或DAMPs,从而触发小胶质细胞的激活和随后的神经炎症级联反应。例如,TLR-4被报道在AD及相关痴呆中扮演重要角色,促进Aβ和NFT的形成或沉积。进一步研究发现,激活的小胶质细胞表达一系列受体,其中TREM2信号对于维持小胶质细胞代谢至关重要,其缺乏会损害小胶质细胞的存活、迁移和吞噬活性。同时,PRRs与Aβ结合后可引发胶质细胞激活,增加激活的小胶质细胞数量及其密度,并通过NF-κB的核转位促使TNF-α和其他促炎细胞因子的生成,导致神经炎症(图3)。
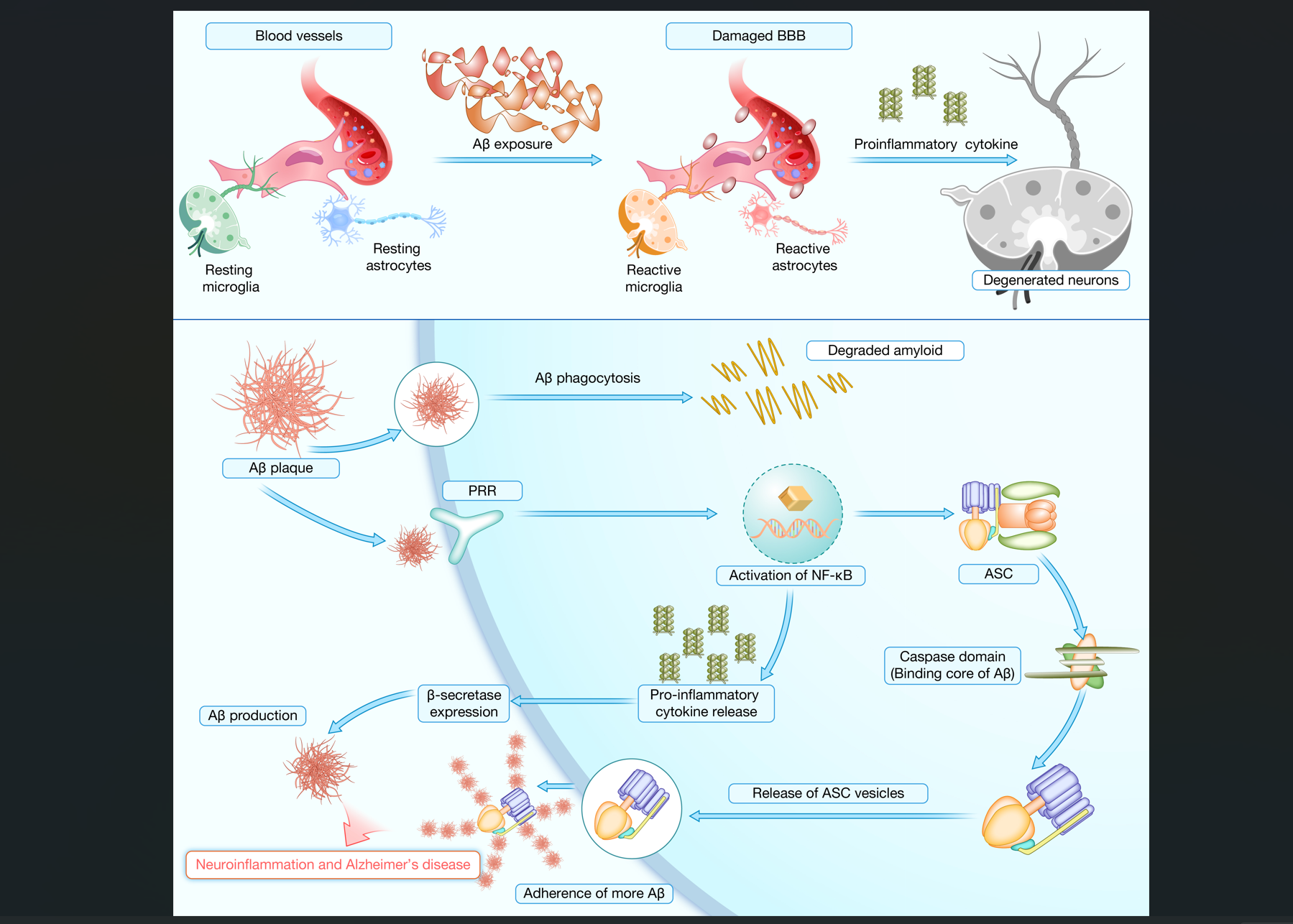
图3 Aβ在AD中的病理作用
Tau蛋白、Aβ与AD的关系
Aβ是老年斑的主要成分,由淀粉样前体蛋白分解产生,而过度磷酸化的tau蛋白则形成NFT,二者均参与神经退行性病变过程。激活的小胶质细胞与聚集的Aβ42构成的老年斑有关,且随着小胶质细胞的激活和AD进展,老年斑形成增多(图4)。此外,氧化应激介导的神经毒性可能增加Aβ、tau及其超磷酸化的积累。小胶质细胞的激活加速了ROS的释放,促进了淀粉样斑块的形成。尽管小胶质细胞与Aβ之间的联系早在上世纪90年代就被发现,但对于它们之间原子层面的相互作用仍不完全清楚。相比之下,关于tau和小胶质细胞的研究较少,但tau蛋白通常与微管结合以稳定其结构,而在AD中,tau脱离微管后变得不可溶,形成NFT,进一步激发周围反应性小胶质细胞的活化。
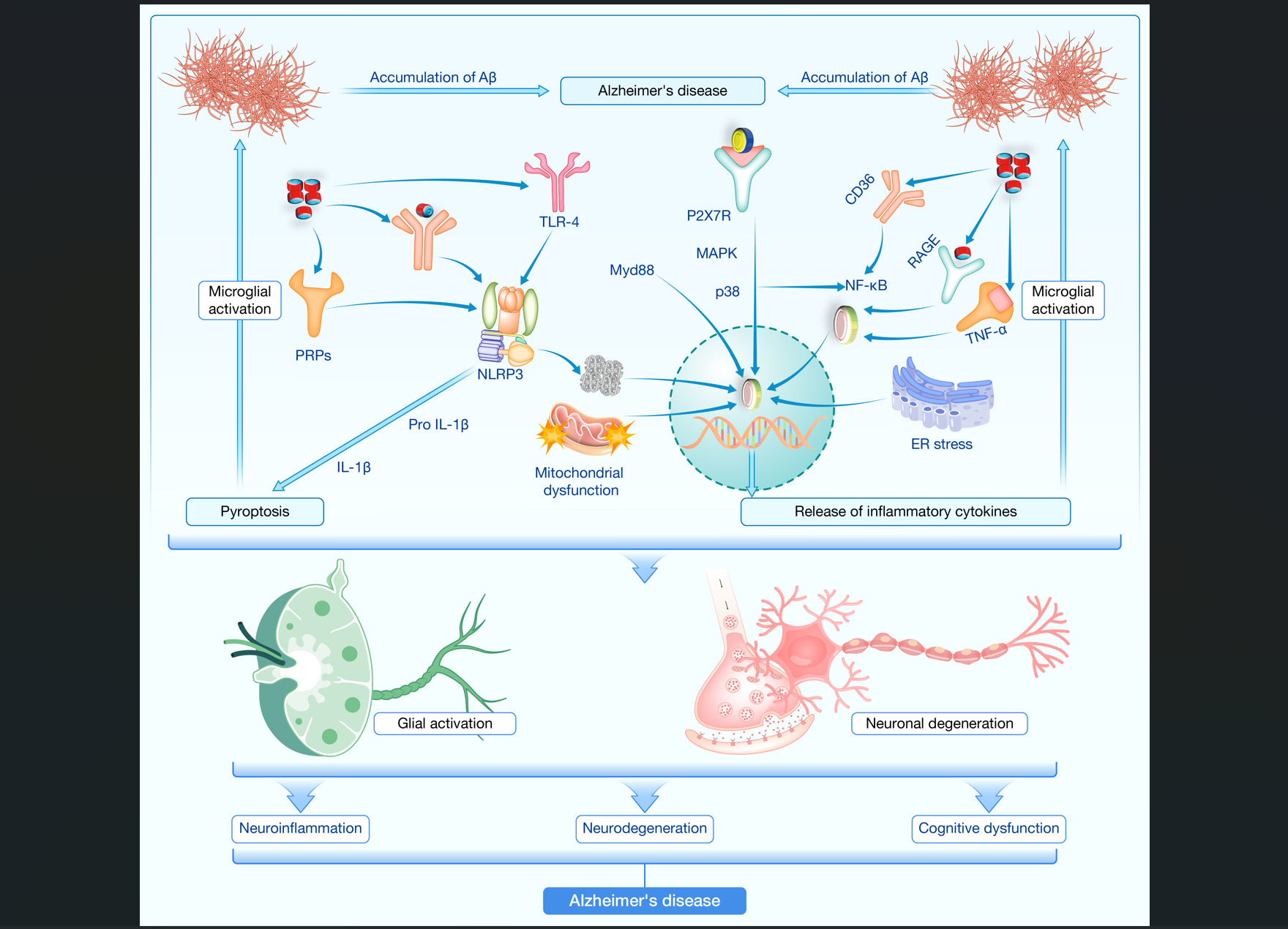
图4 AD中的神经炎症
神经炎症与小胶质细胞
随着细胞与分子研究的深入,人们发现神经炎症在正常生理状态下是维持大脑稳态的基本机制,而在病理状态下则由星形胶质细胞和小胶质细胞释放促炎因子(如TNF-α、IL-1β、ROS等)引发。当血脑屏障受损时,外周免疫细胞进入中枢神经系统,进一步加剧炎症反应,形成慢性过程。这种炎症状态一旦启动,即使体内释放抗炎因子也难以控制,最终可能导致突触功能障碍、神经毒性及认知下降。研究表明,活化的小胶质细胞和星形胶质细胞在AD中既可能具有清除蛋白聚集物的保护作用,也可能因过度激活而促进神经炎症,尤其是在AD患者死后脑组织中已发现大量促炎性星形胶质细胞的存在。
神经炎症对痴呆症的影响及治疗策略
越来越多的证据表明,神经炎症不仅影响多种神经系统疾病(如抑郁症、多发性硬化),更是AD及相关痴呆的重要风险因素之一。据世界卫生组织统计,目前全球约有5500万人患有痴呆,预计到2050年将超过1.4亿,其中AD占比超过70%。Aβ沉积、tau蛋白病变以及神经炎症三者之间相互促进,构成AD发病的核心机制。特别是tau蛋白在GSK-3β和MAPK等激酶作用下发生过度磷酸化后,会引发NFT形成,并进一步诱导CXCL3和NLRP3炎症小体的激活,从而加剧神经炎症和认知衰退(图5)。此外,小胶质细胞相关基因突变也被证实与神经炎症和痴呆密切相关。鉴于此,针对神经炎症和小胶质细胞激活的治疗策略正在被广泛探索,包括Urolithin A、维生素E、吲哚美辛等药物,均显示出一定的抗炎和神经保护潜力。
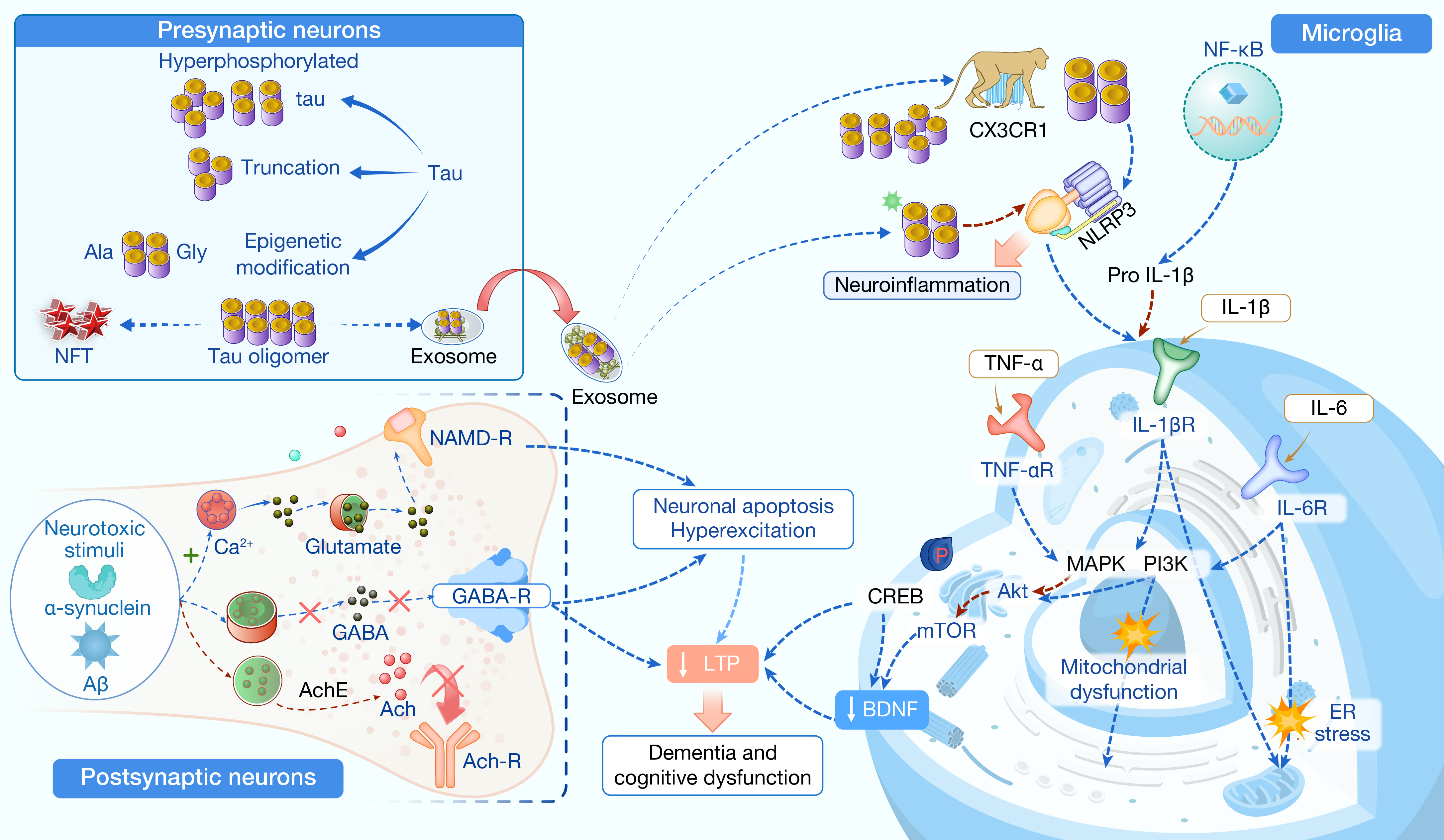
图5 AD中的神经炎症与痴呆
小胶质细胞对认知功能的影响机制
研究表明, AD中的认知障碍主要与海马体突触密度减少而非Aβ或tau损伤有关。可溶性β寡聚体通过改变突触结构损害认知功能,而神经干细胞可能对抗这种有害影响。在AD模型中,小胶质细胞促炎因子增加、分支减少,导致慢性炎症和认知衰退。随着小胶质细胞监测能力减弱,认知功能逐渐恶化。免疫基因表达和小胶质细胞数量的变化也影响衰老过程中的认知表现,如晚发性AD患者脑脊液中高水平的前颗粒蛋白(PGRN)与较差的认知表现相关。TREM2及其可溶形式与PGRN呈正相关,调节其信号有助于控制认知障碍的发展。此外,碎片化睡眠加速衰老并诱发小胶质细胞活化及认知功能障碍,而缩醛磷脂等物质可通过抑制炎症途径改善轻度认知障碍。
小胶质细胞受体作为潜在靶点
小胶质细胞受体在调控脑内炎症免疫反应中的关键作用使其成为AD治疗的潜在靶点。TREM2、NLRP3、RAGE和SR-AI等受体不仅参与tau蛋白聚集的清除,还调节促炎因子的释放,其表达水平与AD病理进展和症状表现密切相关。研究显示,补体受体CR1与晚发性AD有关,靶向CR1等补体受体有望阻止疾病向更严重的神经退行阶段发展。此外,代谢型谷氨酸受体(mGluR)中的III型亚型激活具有神经保护作用,其刺激有助于抑制神经元死亡;而II型亚型则促进神经毒性小胶质细胞的活化。GABA通过激活小胶质细胞上的受体减少促炎因子释放,显示出抗炎潜力。嘌呤能受体、A2A受体和CB2受体也参与调控小胶质细胞活性,例如激活CB2受体可抑制由纤维状Aβ引发的小胶质细胞过度活化及其导致的神经毒性。同时,通过调节MAPK信号通路,大麻素类物质也能减轻炎症反应,展现神经保护作用。由此可见,针对小胶质细胞受体的干预可能为延缓AD进展提供新的治疗策略。
AD的非药物干预
尽管已有多种针对AD的药物处于不同研发阶段,但目前仍缺乏有效的治疗手段,因此非药物干预逐渐成为管理AD的重要策略之一。常见的非药物干预包括光生物调节、益生菌使用、粪菌移植、深部脑刺激、针灸、音乐疗法、认知训练以及生活方式调整等。其中,光生物调节(PBM)通过特定波长(如660–810 nm)作用于线粒体细胞色素c氧化酶,激活信号通路,提升ATP生成,改善线粒体功能,从而发挥抗氧化、抗炎及神经保护作用。动物实验表明,PBM可有效减少Aβ沉积并改善认知功能。此外,运动作为生活方式干预的核心部分,不仅通过上调PGC1α表达降低促炎因子水平,还促进Irisin分泌,增强血脑屏障通透性和BDNF表达,从而改善认知能力。
饮食调节与肠道菌群干预也显示出良好前景。高胆固醇和脂质代谢紊乱是AD的风险因素,而益生菌和粪菌移植(FMT)可通过恢复肠道微生态平衡,减少炎症反应,降低COX-2和CD11b表达,改善突触可塑性。临床试验显示,益生菌干预能显著降低氧化应激标志物CRP和MDA水平。在侵入性干预方面,深部脑刺激(DBS)被证实可减少Aβ沉积、抑制神经炎症,并在动物模型中改善认知功能。非侵入性脑刺激技术如经颅直流电刺激(tDCS)和重复经颅磁刺激(rTMS)也在多项研究中展现出潜在疗效。总体而言,非药物干预副作用少、安全性高,被视为AD综合管理中的重要补充,但仍需进一步机制研究和长期临床验证以确立其疗效。
AD在临床上的治疗策略
目前,用于治疗AD的药物寥寥无几,如多奈哌齐、加兰他敏、美金刚、利斯的明、阿杜卡单抗和仑卡奈单抗等。这些药物主要为胆碱酯酶抑制剂或N-甲基-D-天冬氨酸(NMDA)受体拮抗剂,仅能缓解症状,并不能治愈疾病。此外,单克隆抗体也未能显示令人鼓舞的结果,因此研究转向了新的靶点,例如tau蛋白及神经炎症相关的靶点。根据最新发布的报告,在143种AD研发管线药物中,超过80%旨在改变疾病的进程,其中约15%-16%针对Aβ,10%针对tau蛋白,另有大约20%专注于神经炎症。
在探索新靶点方面,GSK-3β或MAPK等激酶因其在tau过度磷酸化中的作用而受到关注,其抑制剂锂盐和替德格鲁昔布曾被研究用于对抗AD及其认知衰退的影响。然而,尽管锂盐长期使用可显著减缓认知功能衰退,但其有效剂量引发了关于糖尿病、心律失常、甲状腺及肾功能的安全性和耐受性担忧。替德格鲁昔布在动物模型中显示出减少tau蛋白过度磷酸化的潜力,但在临床试验中对轻度至中度AD患者未见明显疗效。另外,O-GlcNAcase (OGA)抑制剂如LY3372689、ASN90、ASN51正在进行II/III期临床试验,因其I期试验已显示出良好的安全性和耐受性。除了上述药物,还有针对tau蛋白翻译后修饰的疫苗和药物开发策略,AADvac1和ACI-35/ACI-35.030是目前正在临床试验中的两种疫苗。此外,为了应对由神经炎症导致的小胶质细胞激活和AD相关认知能力下降的问题,目前有多种药物正在探索之中,包括p38α抑制剂如尼洛替尼和β-雄烯二醇。值得注意的是,文中提到的药物靶向tau病变情况汇总于表1,提供了进一步的研究视角和数据支持。
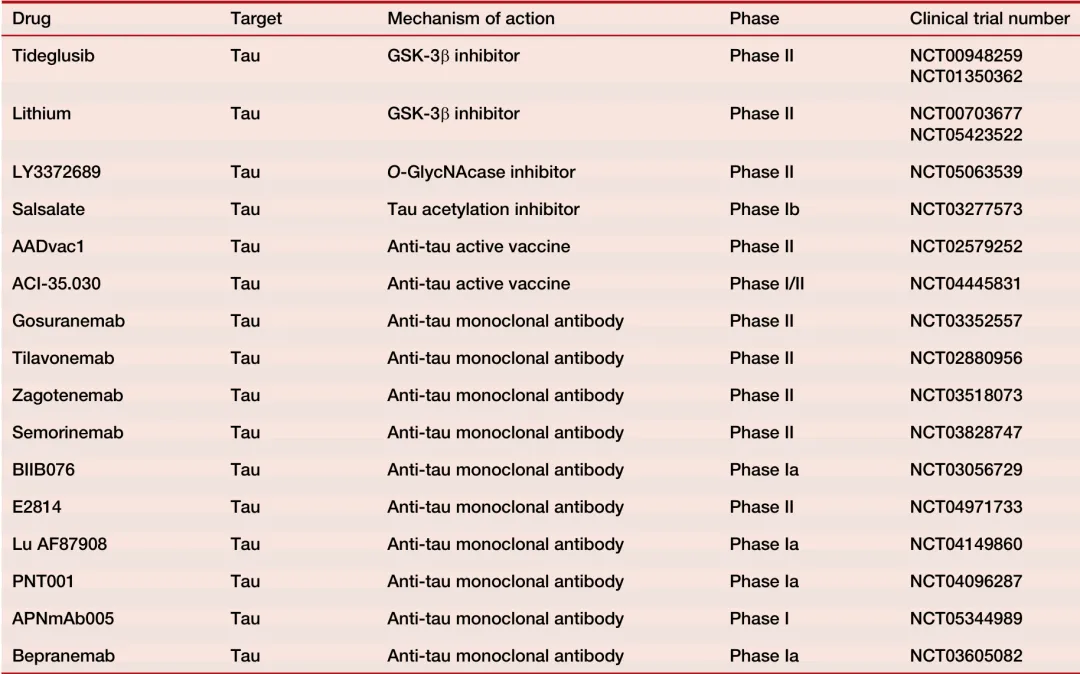
表1 处于临床试验各阶段的药物
针对AD中小胶质细胞激活的药物临床开发
针对AD的治疗策略主要集中在降低Aβ水平,但迄今为止效果不佳。例如,口服β-分泌酶1(BACE-1)抑制剂未能减缓轻至中度AD患者的认知或功能衰退。此外,多项临床试验确认其无法彻底清除患者脑内的不同形式Aβ和tau蛋白,因此仅去除斑块不足以作为症状性AD的有效治疗手段,但仍可能作为一种预防措施。鉴于此,通过靶向小胶质细胞来调节神经炎症成为一种潜在且可能更有效的治疗方法。多种药物如Masitinib、Simufilam、ALZ-801等正在开发中,其中一些已经进入II期或III期临床试验阶段。特别是针对TREM2的小分子抑制剂AL002和抗体TAK-920/DNL919,旨在抑制小胶质细胞活化并改善AD病情(表2)。
进一步的研究探索了其他抗炎药物在AD中的应用,包括选择性CB2受体激动剂、Epidiolex(纯度99%的大麻二酚提取物)、JNJ-40346527(CSF1R抑制剂)以及OAB-14等。这些药物分别通过不同的机制减轻神经炎症、促进Aβ清除及保护神经元。例如,JNJ-40346527能够减少微胶质细胞增殖和炎症因子表达,而OAB-14则刺激胰岛素降解酶(IDE)和内肽酶(NEP)的表达,增强微胶质细胞吞噬作用。此外,Nilvadipine、pioglitazone、NE3107等药物也因其抗炎特性被纳入不同阶段的临床试验。值得注意的是,Canakinumab作为一种IL-1β中和抗体,在一项预计于2026年结束的II期试验中评估其对AD的效果。这些研究不仅为AD治疗提供了新视角,而且强调了调节CB2受体及其他炎症途径的重要性,为进一步理解炎症与微胶质细胞激活的关系奠定了基础。综上所述,尽管许多药物仍处于研发初期,但它们为未来AD的非Aβ靶向治疗带来了希望。
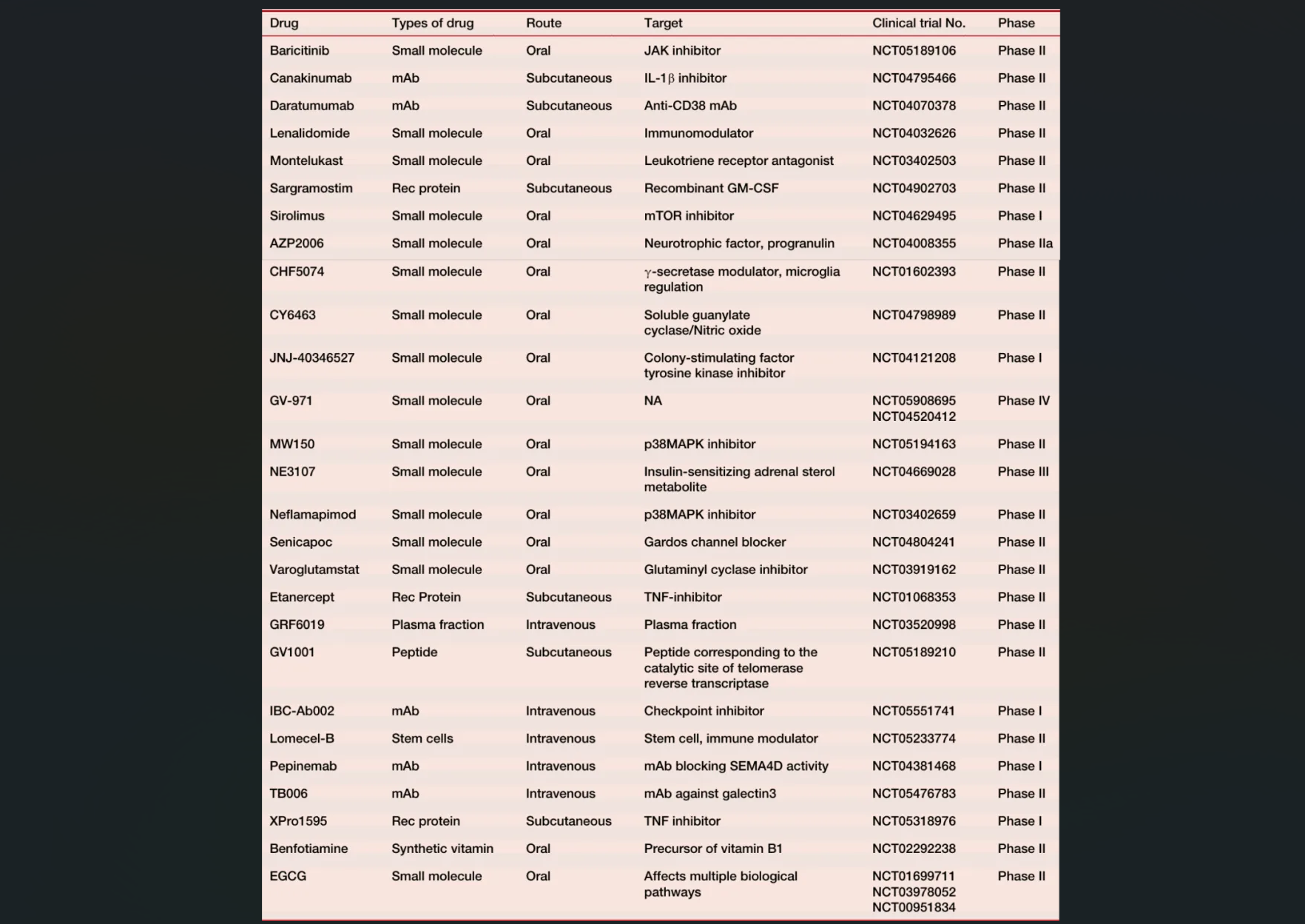
表2 目前正在进行神经炎症临床试验的抗炎药物
总结与展望
过去几十年中,基于细胞和分子的研究,AD的多维度致病特征已被充分认识,多种信号分子和通路被确定与AD发病相关。神经炎症被认为是导致痴呆和认知衰退的主要因素之一。尽管近五年来美国食品药品监督管理局(FDA)批准了若干免疫治疗药物,且更多药物正在审批过程中,但大多数获批药物的疗效和安全性仍存在争议。这是因为这些药物主要针对Aβ,而Aβ并非AD发病的核心因素。
除Aβ外,tau蛋白也被作为管理和治疗AD的一个靶点进行研究,不过目前尚无FDA批准的针对tau蛋白的药物,这为药物研发提供了进一步探索的空间。近期,小胶质细胞-星形胶质细胞相互作用、小胶质细胞活化及线粒体功能障碍在AD病理中的角色也受到了关注。值得注意的是,小胶质细胞的激活被认为是AD发病及其并发症的关键因素。鉴于此,以小胶质细胞活化为目标的抗炎药物得到了研究。然而,结论是单一靶点药物可能不足以应对AD及其相关的认知功能障碍,具有多重机制的药物才是更优的选择。
*此微信稿为翻译稿,如有歧义请以英文原文为准。
作者简介
Mohammad Kashif Iqubal 博士后
通讯作者
机构:得克萨斯A&M大学
研究方向:基于纳米制剂的皮肤癌、皮肤疾病和神经退行性疾病的治疗
Ashif Iqubal 研究员
通讯作者
机构:印度新德里Jamia Hamdard大学
研究方向:抗癌药物诱导的多器官毒性和辅助治疗
引用格式:Ghimire A, Rehman SA, Subhani A, et al. Mechanism of microglia-mediated neuroinflammation, associated cognitive dysfunction, and therapeutic updates in Alzheimer's disease. hLife 2025; 3: 64–81.
期刊简介
hLife 由高福院士、董晨院士和Jules A. Hoffmann教授(2011诺奖获得者)领衔,是中国科学院微生物研究所主办,中国生物工程学会,浙江大学陈廷骅大健康学院,西湖大学医学院,上海市免疫治疗创新研究院和广州霍夫曼免疫研究所联合支持,与国际出版商爱思唯尔合作的健康科学领域综合性英文期刊。
hLife 聚焦健康科学领域的前沿进展,旨在促进基础研究与临床应用的融合发展。期刊发表与医学相关各研究领域最新成果,学科领域包括(但不限于)病原生物学、流行病学、生理学、免疫学、结构生物学、疾病监测、肿瘤、药物、疫苗和健康政策等。
hLife是一本金色开放获取期刊,月刊出版;2022年成功入选“中国科技期刊卓越行动计划高起点新刊”;2023年11月正式创刊;2024年5月被DOAJ收录;2024年8月被Scopus收录。
2026年前hLife接收的稿件免收文章处理费(APC)。
期刊网址:
https://www.sciencedirect.com/journal/hlife
https://wap.sciencenet.cn/blog-3552961-1492340.html
上一篇:[转载]hLife | 美国国家癌症研究所郑志明和Vladimir Majerciak研究团队破译质粒DNA转染激活双重免疫应答
下一篇:[转载]hLife | “损伤管控”+“刺激修复”策略:孤独症治疗新观点