博文
代谢学人——Cell Metabolism:松开代谢刹车,肥胖一去不返
||
撰文 | 李欣茜 张婷 仲银召 郑宇含 张彦康 李雨
编辑 | 孟美瑶
校对 | 张婷

研究背景
肥胖的患病率逐年升高,预计到2030年,肥胖将影响全球超过10亿成年人,这将对医疗系统和社会经济产生巨大压力。肥胖相关并发症主要包括2型糖尿病、心血管疾病、癌症和呼吸系统疾病等,这些并发症与脂肪组织功能失调息息相关。有报道指出,肥胖往往伴随着脂肪组织中线粒体含量减少和氧化代谢降低,这提示线粒体可能在脂肪组织的脂质代谢、胰岛素信号传导和能量消耗等方面发挥重要作用。棕色脂肪细胞含有大量线粒体,能迅速氧化燃料,从而驱动无效化学循环,将化学能转化为热量。此外,米色脂肪细胞也是一种产热脂肪细胞,能在受到寒冷或β3-肾上腺素能激动剂刺激时被激活,从而具有和棕色脂肪细胞相似的表型和产热能力。棕色脂肪细胞和米色脂肪细胞在维持体温和改善代谢健康方面发挥着重要作用。
在肥胖状态下,脂肪组织中免疫细胞(如促炎性巨噬细胞和T细胞)显著增加,并可以通过释放细胞因子来影响脂肪细胞的线粒体功能。例如,肿瘤坏死因子α(TNF-α)、白细胞介素-1β(IL-1β)和干扰素(IFN)都能抑制线粒体功能和产热。此外,脂肪细胞中的先天免疫传感器,包括脂多糖受体Toll样受体4(TLR4)和cGAS-STING,可能部分通过激活干扰素调节因子3(IRF3)来抑制肥胖中的产热。因此,探究炎症信号影响线粒体功能的机制可能为在炎症存在的情况下维持氧化代谢提供新方法,同时能为肥胖的病理生理学提供新见解。
转录共激活因子PGC1α是几乎所有组织中线粒体生物合成和氧化代谢的主要调控因子。PGC1α能协调线粒体生物合成和调控产热相关基因表达,因此PGC1α是脂肪组织中氧化代谢的核心,其基因敲除会抑制产热并导致胰岛素抵抗。尽管PGC1α的表达主要受β-肾上腺素能信号途径的转录调控,但近期的研究发现针对Ppargc1a mRNA的转录后调控也十分重要,例如,本文研究团队此前发现Ppargc1a的5’UTR区域含有一个负调控其翻译的上游开放阅读框(uORF)(小编注:uORF是mRNA的5'端非翻译区(5' UTR)中的一类开放阅读框,uORF的起始密码子通常位于主要开放阅读框(mORF,即CDS区)的起始密码子上游,可以影响mORF的翻译效率。在真核生物转录组中大约一半mRNA的5' UTR中含有uORF。uORF通常以AUG作为起始密码子,少数uORF可以编码小肽,协助植物抵御干旱、高盐胁迫。更多的uORF功能是调节mORF翻译。具体而言:uORF可以通过竞争性结合核糖体,阻止核糖体与mORF的起始密码子结合,从而抑制翻译起始。核糖体翻译含有uORF的mRNA时可能发生的三种情况:渗漏扫描、重新起始、停滞/解聚。渗漏扫描指核糖体会跳过uORF区域,与mORF的起始密码子结合,正常翻译;重新起始指核糖体在翻译完uORF后,重新定位到mORF的起始密码子并开始翻译,对mORF的翻译具有一定的抑制;停滞/解聚指核糖体在翻译uORF时发生停滞或解聚,阻止核糖体翻译mORF。总之,uORF对mORF翻译起始总体表现为抑制作用。在这里提到的是本文团队在2019年发表在CM上的文章,他们发现Ppargc1a的5’UTR区域含有一个负调控PGC1α翻译的Ppargc1a uORF),其可以调控Ppargc1a翻译,从而在多个组织中调控氧化代谢。
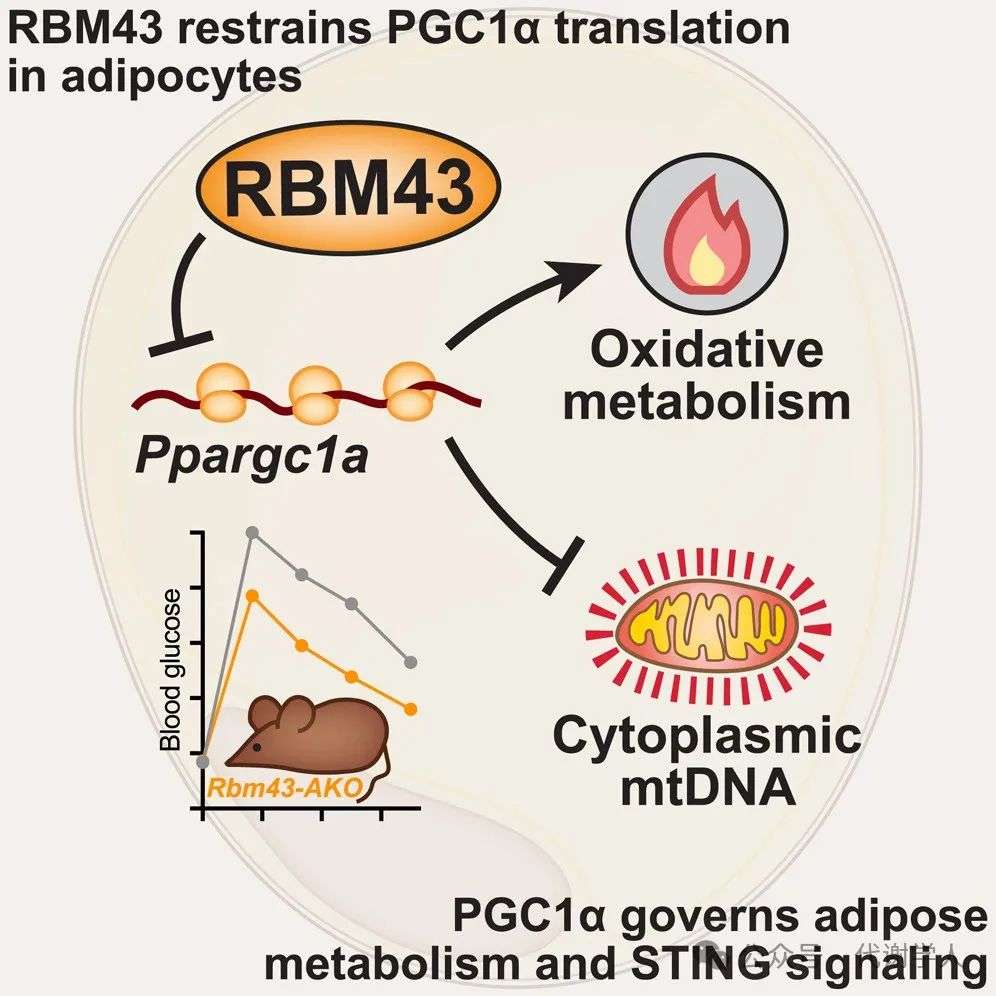
在本篇研究中,研究人员发现RNA结合蛋白RBM43通过抑制PGC1α的翻译,负调控线粒体生物合成和氧化代谢。在肥胖状态下,RBM43敲除能够改善代谢功能,减少炎症反应,并抑制cGAS-STING信号通路。这一发现揭示了RBM43在调节脂肪细胞代谢和炎症反应中的重要作用,为理解肥胖相关代谢紊乱的病理机制提供了新的视角。

敲黑板啦!
1.RBM43抑制PGC1α翻译从而负调控脂肪细胞线粒体生物合成
2.RBM43受到炎症信号诱导,并介导了TNF-α对代谢的负面影响
3.脂肪细胞RBM43介导肥胖下的脂肪炎症和葡萄糖不耐受
4.抑制PGC1α导致细胞质mtDNA积累和cGAS-STING信号传导激活
研究结果
1.RNA结合蛋白RBM43负调控PGC1α翻译和氧化代谢
为了鉴定氧化代谢的转录后调控因子,研究人员首先在公开数据集中筛选与脂肪产热(涉及线粒体含量和功能重塑)相关的RNA结合蛋白。研究人员关注的候选基因包括:(1)RNA结合蛋白或核糖核蛋白复合物;(2)优先在白色或产热(棕色或米色)脂肪细胞中表达;(3)在棕色和腹股沟白色脂肪细胞中并受冷暴露调节。在排除细胞存活所必需的或定位于线粒体的蛋白产物后,得到25个候选蛋白(图S1A)。研究人员针对其分别构建siRNA,并在原代白色脂肪细胞中探究其对PGC1α蛋白:mRNA比率的影响(图S1B),结果发现Rbm43的敲减影响最显著。与产热脂肪细胞相比,Rbm43更倾向于在白色脂肪细胞中表达,且其会受冷暴露刺激而减少表达。Rbm43编码一个39 kDa的蛋白质,其具有一个N端RNA识别基序,有研究表明其与参与肝细胞癌进展的mRNA转录后抑制有关。
接下来研究人员设计两种不同的siRNA来敲减Rbm43,发现二者都能提高PGC1α蛋白:mRNA的比率(都约1.9倍)(图1A和1B),且这一效应未伴有PGC1a蛋白半衰期的变化(在放线菌酮处理期间测定)(图1C)。此外,Rbm43敲低的效应在使用异丙肾上腺素诱导PGC1α转录期间依旧存在(图S1C),表明Rbm43对PGC1α的调控可能是影响蛋白翻译,而非转录调控。
为了更直观的评估RBM43在PGC1α翻译中的作用,研究人员从表达GFP或RBM43的HEK293细胞中制备了体外翻译提取物,并在体外转录出含有PGC1α UTR(非翻译区)序列的萤光素酶报告mRNA。如图1D所示,RBM43降低了含有PGC1α 5’ UTR的PGC1α-nanoLuc mRNA的翻译。RBM43对PGC1α翻译调控不需要PGC1α uORF起始密码子,这表明RBM43不是通过控制核糖体与该uORF的结合来发挥作用(图1D)。
接下来,研究人员在原代脂肪细胞中敲减Rbm43,发现其敲减后可上调脂肪酸氧化、产热和线粒体功能相关基因的表达水平(图1F),但对脂肪生成没有影响,脂肪生成转录调节因子的表达也无明显变化(图1E和S1D)。根据氧耗量测定结果,脂肪细胞产热和氧化代谢相关基因表达的上调与线粒体总呼吸和解耦联呼吸增加相关(图1G)。因此,研究人员断定RBM43是脂肪细胞中PGC1α和氧化代谢的转录后负调节因子。
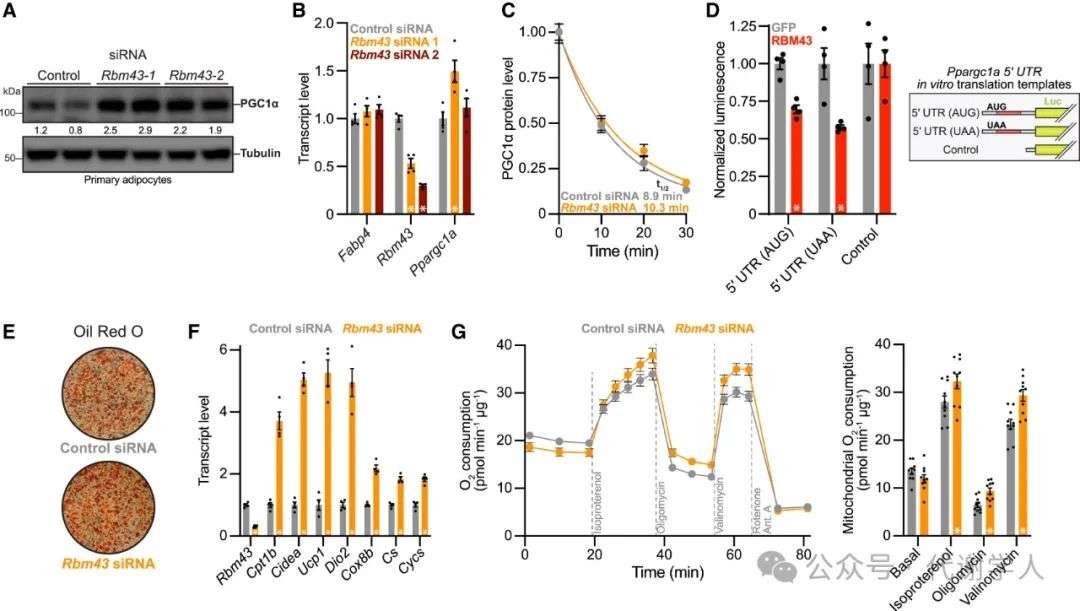
图1.在原代脂肪细胞中敲减Rbm43能提高PGC1α蛋白水平并增强线粒体呼吸
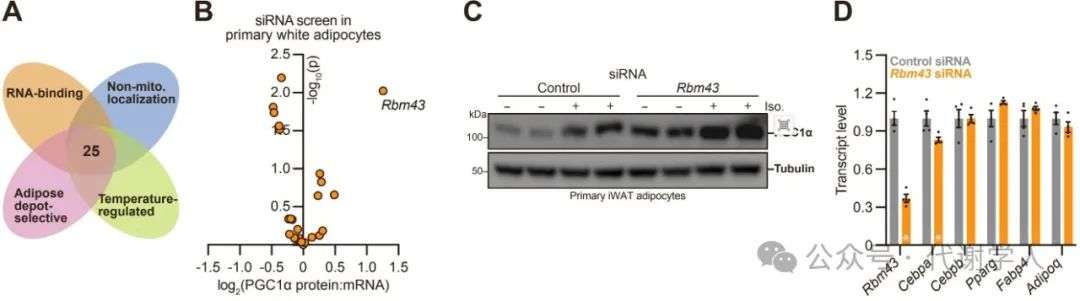
图S1.脂肪细胞氧化代谢的候选转录后调控因子
2.RBM43在白色脂肪中选择性表达,并受炎症信号诱导
接下来,研究人员进一步探究了RBM43 mRNA的表达状况。研究人员在22℃条件下饲养小鼠的所有组织中均检测到Rbm43的表达,其中白色脂肪组织(WAT)(包括腹股沟WAT(iWAT)和内脏脂肪组织(VAT))和脾脏中的Rbm43表达水平最高(图S2A),而棕色脂肪组织(BAT)中的Rbm43水平最低,比WAT低80%(图2A和S2A)。接下来研究人员对公开的翻译核糖体亲和纯化(TRAP)数据(小编注:TRAP是利用在特定细胞类型中表达的 GFP 标记的核糖体蛋白,从而允许在不分离组织的情况下,从复杂组织中的靶细胞中分离出核糖体结合的mRNA)进行分析,证实Rbm43在白色脂肪中特异性表达,而非产热脂肪(图2B)。
当小鼠暴露于低温(4℃)以诱导脂肪产热时, VAT、iWAT和BAT中Rbm43的表达水平均降低(下降幅度约73%到83%)(图2C和2D)。在aP2-Prdm16转基因小鼠的iWAT和VAT脂肪细胞中,Rbm43的表达也较低,并伴随产热米色脂肪细胞数量增加(图S2B)。由于肾上腺素能信号驱动产热程序的许多方面,研究人员评估了其在原代脂肪细胞中对Rbm43的影响。结果发现,肾上腺素能激动剂和增加细胞cAMP的其他药物可降低Rbm43表达(图2E)。有研究表明,Rbm43基因的启动子区域存在IRFs和NF-κB的潜在结合位点,提示炎症信号可能参与Rbm43的表达调控。于是研究人员用IFN-α、IFN-γ、TNF-α或IL-1β处理原代脂肪细胞,发现Rbm43表达增加,其中IFN-α诱导的效应最大(9倍)(图2F)。IFN应答不仅可由IFN受体启动,也可由固有免疫系统的传感器启动,如TLR、RIG-I和cGAS STING。因此研究人员用脂多糖(LPS,TLR4配体)、聚肌苷酸(poly-I:C,能激活TLR3以及RNA受体RIG-I和MDA5)、diABZI(STING激活剂)和cGAMP(STING激活剂)分别处理原代脂肪细胞,发现其均可诱导Rbm43表达(图2G)。其中diABZI、cGAMP效果更好,使Rbm43 mRNA上调了15倍(图2G)。
RBM43在人类中的调节作用与小鼠中相同。与棕色脂肪细胞相比,RBM43在人类白色脂肪细胞中选择性表达。另外在腺苷酸环化酶激活剂Forsklin处理后,白色脂肪细胞中cAMP信号传导增强,RBM43表达被下调(图2H)。研究人员用促炎细胞因子或上述四种激活先天免疫信号传导的试剂处理人脂肪细胞,发现同样能上调RBM43的表达。由于TNF-α能通过RELA(p65)来激活NF-κB信号通路,研究人员在人脂肪细胞中进行验证,发现敲除p65后,TNF-α上调RBM43的趋势被显著抑制,表明TNF-α对其的调节需要p65(图2I、2J和S2C)。上述小鼠和人类细胞中的结果表明,Rbm43受到肾上腺素能信号的负调控,受到炎性细胞因子和先天免疫受体的正调控。
最后,研究人员探究了Rbm43的缺失是否能挽救TNF-α引起的线粒体损伤。研究人员用TNF-α处理原代脂肪细胞24小时,发现TNF-α可显著降低线粒体酶编码基因的表达。在该细胞模型中,Rbm43敲减逆转了TNF-α对九种三羧酸(TCA)循环酶mRNAs中八种的下调,促使其表达平均增加42%(图S2D)。另外Rbm43敲减还增加了乌头酶2(ACO2)和氧化磷酸化复合物亚基ATP5A和SDHB的蛋白水平(图S2E和S2F)。值得注意的是,Rbm43敲减显著恢复了TNF-α对脂肪细胞呼吸能力的不利影响(图S2G)。因此,RBM43是TNF-α抑制线粒体基因表达和氧化代谢的部分原因。
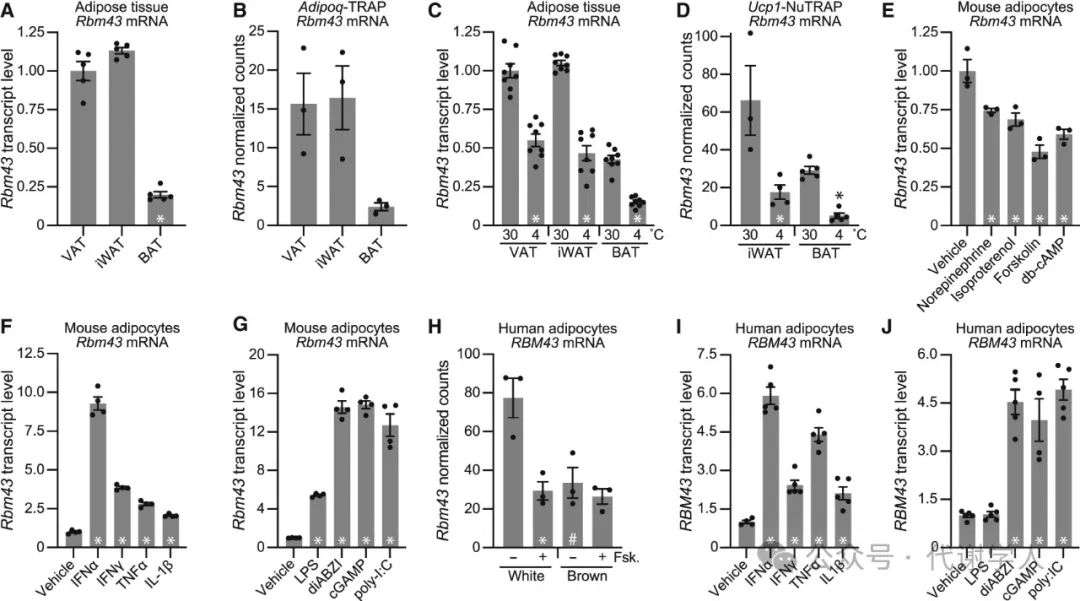
图2.Rbm43受肾上腺素信号和炎性细胞因子的调节
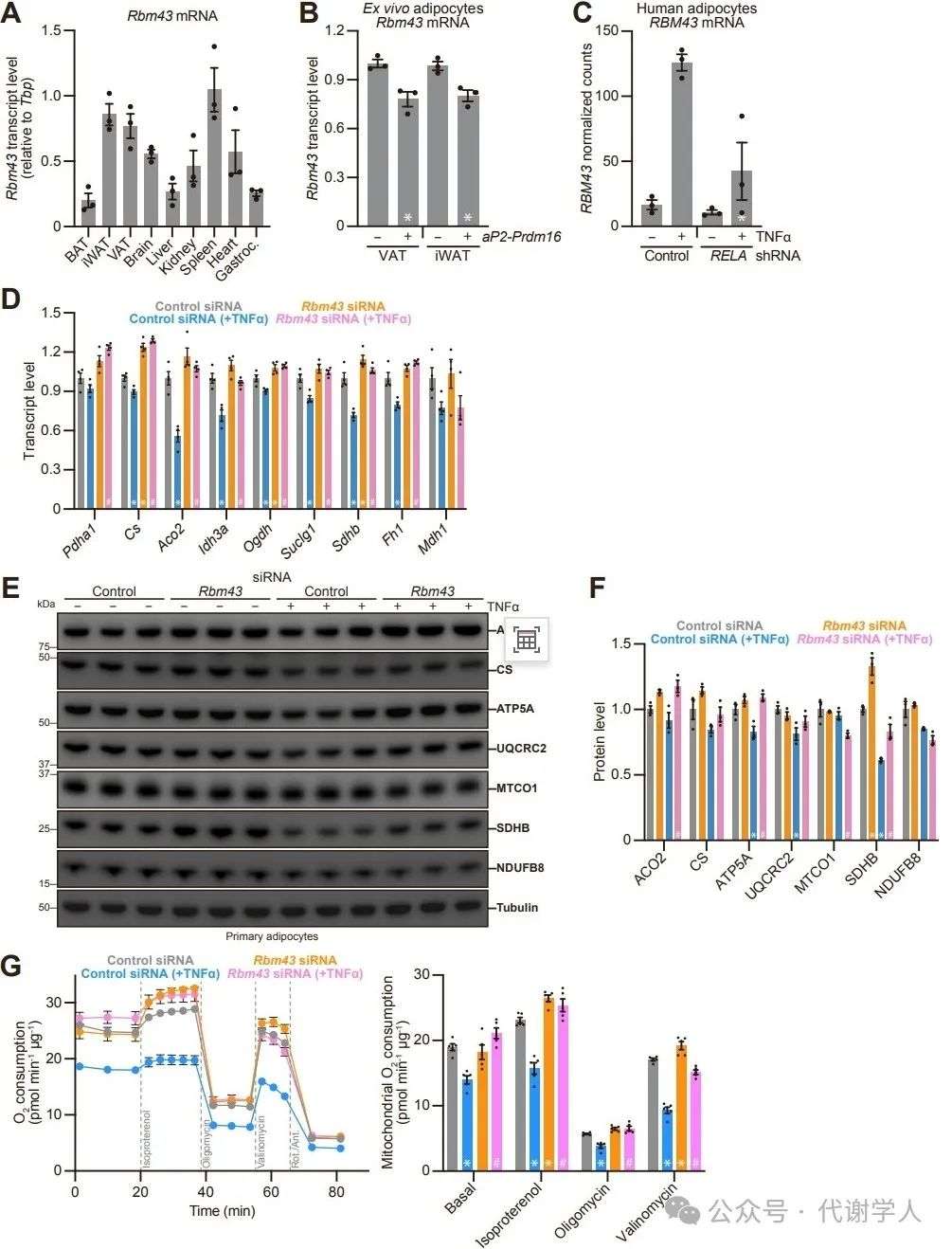
图S2.Rbm43的表达及其在TNF-α诱导的线粒体损伤中的作用
3.RBM43以PGC1α依赖的方式调控氧化代谢基因表达
接下来,研究人员探究了RBM43调控的整体基因表达,以及其对PGC1α的依赖程度。首先,研究人员用腺病毒介导的Cre或GFP(绿色荧光蛋白)来处理原代PGC1αfl/fl脂肪细胞,以条件性敲减PGC1α。尽管GFP的荧光检测显示超过90%的脂肪细胞被成功转导了Cre重组酶,但实际被Cre重组酶敲减的PGC1αfl/fl只有大约50%(图S3A和S3B)。虽然Cre重组酶对PGC1αfl/fl的敲减存在不完全性,但由于PGC1α蛋白在敲减后几乎恢复到基线水平,使得研究人员能够更准确地研究RBM43对PGC1α的特定影响。
随后,研究人员对PGC1α敲减的原代脂肪细胞进行RNA-seq分析,发现Rbm43敲减提高了693个基因的mRNA表达(log2FC>0.3;padj<0.005)(图3A和3B)。氧化磷酸化、脂肪生成和脂肪酸代谢是三个最显著的Hallmark基因集。这些基因集包括氧化磷酸化程序的核心成分,包括PGC1α的直接和间接靶标。基因集富集分析显示,Rbm43敲减显著恢复了PGC1α的正常活性特征(图3C和3D)。以上结果表明,RBM43敲减导致PGC1α活性升高。为了验证PGC1α在Rbm43敲减引起的基因表达变化中的作用,研究人员用Cre重组酶敲减PGC1α,结果发现其敲减逆转了大多数基因的表达上调:492个(71%)基因恢复到接近基线水平(Z>0.67的变化)(图3B)。
与Rbm43敲减后上调的基因相比(693个),下调的基因数量较少(253个)(图3E和3F)。在下调的基因中,上皮间质转化(EMT)、未折叠蛋白反应和炎症反应是富集最显著的基因集。Cre介导的PGC1α敲低仅恢复了RBM43敲低后下调的少数基因(66个,占比26%)(图3F)。然而,值得注意的是,剩余的PGC1α非依赖性基因并没有富集到任何特定的基因集中。总之,这些结果揭示了RBM43主要在以下两个方面调控基因表达,一方面是通过抑制PGC1α来下调氧化代谢相关的基因,另一方面是激活与细胞特性、应激和炎症相关的基因,其中部分基因同样依赖于PGC1α发挥作用。
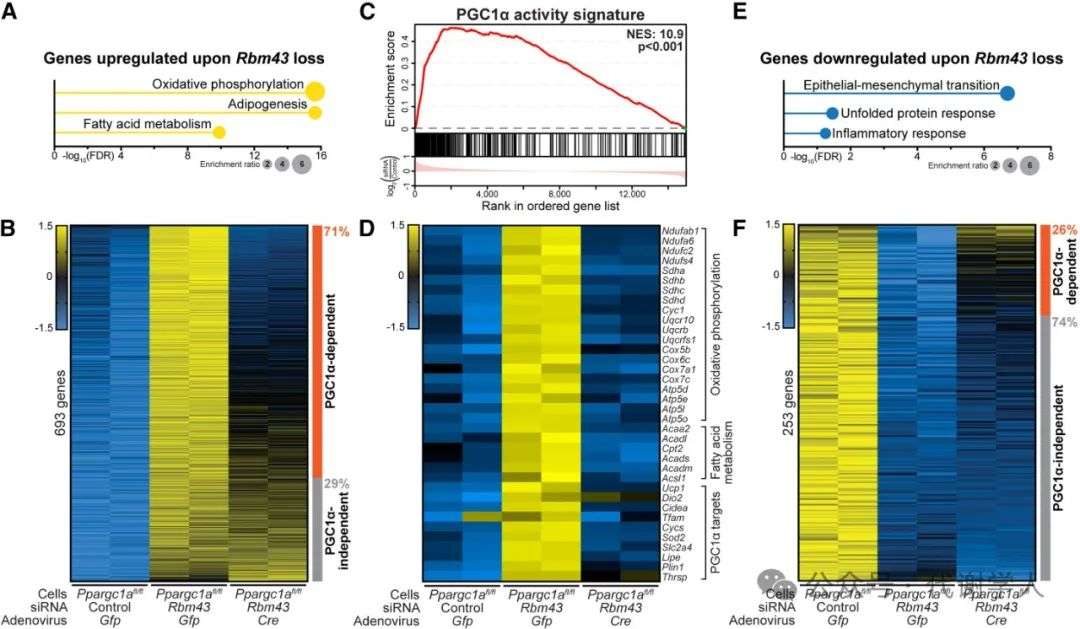
图3.RBM43通过PGC1α控制脂肪细胞氧化磷酸化基因表达程序
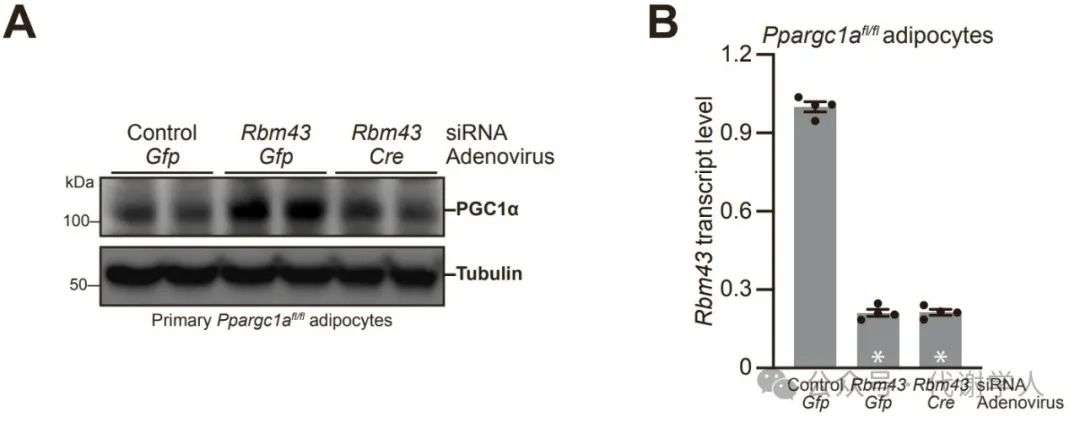
图S3.Cre介导的Ppargc1afl/fl敲减使Rbm43敲减细胞中PGC1α恢复到基线水平
4.脂肪细胞特异性敲除Rbm43可促进PGC1α翻译水平、线粒体生物合成和氧化代谢
接下来,研究人员构建了脂肪细胞特异性敲除Rbm43(Rbm43 AKO)小鼠,Rbm43 AKO小鼠按孟德尔比例出生,无明显异常。且Rbm43 AKO小鼠与同窝Rbm43fl/fl小鼠的棕色和白色脂肪组织的重量没有差异(图S4A和S4B)。
研究人员发现,与Rbm43fl/fl小鼠相比,Rbm43 AKO小鼠iWAT组织中PGC1α蛋白表达显著上调,而Ppargc1α基因表达无明显差异,这再次表明Rbm43可能参与调控Ppargc1α的翻译水平(图4A和4B)。随后,研究人员利用环己亚胺处理Rbm43 AKO原代脂肪细胞,以固定核糖体与mRNA的结合,随后裂解细胞并进行蔗糖密度梯度离心,其中单体mRNA处于低密度部分,而与核糖体结合的mRNA处于高密度部分。研究人员发现,与Rbm43fl/fl原代脂肪细胞相比,Rbm43 AKO原代脂肪细胞中Ppargc1α mRNA在蔗糖高密度部分的含量更高,而Retn mRNA(作为对照)无显著变化(图4C),表明Rbm43缺失促进了Ppargc1α的翻译水平。
为了更广泛地评估RBM43在脂肪细胞中的调控作用,研究人员对Rbm43 AKO原代脂肪细胞进行蛋白质组学分析,结果显示Rbm43缺失后显著上调了氧化磷酸化、脂肪生成和脂肪酸代谢途径,下调了EMT和炎症信号(图4D)。正如预期那样,Rbm43缺失后显著激活了PGC1α活性特征,包括氧化磷酸化和脂肪产热相关基因和蛋白(图4E-4I)(小编注:PGC1a activity signature是对Rbm43 AKO原代脂肪细胞的RNA-seq进行GSEA分析,其中包括氧化磷酸化、脂肪酸氧化和PGC1α靶点相关基因,这些是文献已报道的PGC1α的直接或间接靶点)。这些基因和蛋白在Rbm43 AKO小鼠的VAT中同样上调(图S4C-S4E)。在BAT中,Rbm43对产热和氧化代谢相关基因表达无明显影响(图S4G)。
随后,研究人员检测了RBM43缺失对脂肪氧化代谢能力的影响,结果显示,Rbm43缺失显著提高了iWAT组织的OCR(耗氧量)水平,而对VAT无显著影响(图4J和S4F)。接下来,研究人员利用代谢笼监测Rbm43 AKO小鼠的机体能量消耗水平,结果显示,Rbm43 AKO小鼠机体能量消耗与Rbm43fl/fl小鼠无显著差异,而CL-316,243处理显著提高了Rbm43 AKO小鼠的能量消耗(图4K)。总之,这些结果表明Rbm43可抑制白色脂肪中线粒体的生物发生和氧化代谢水平。
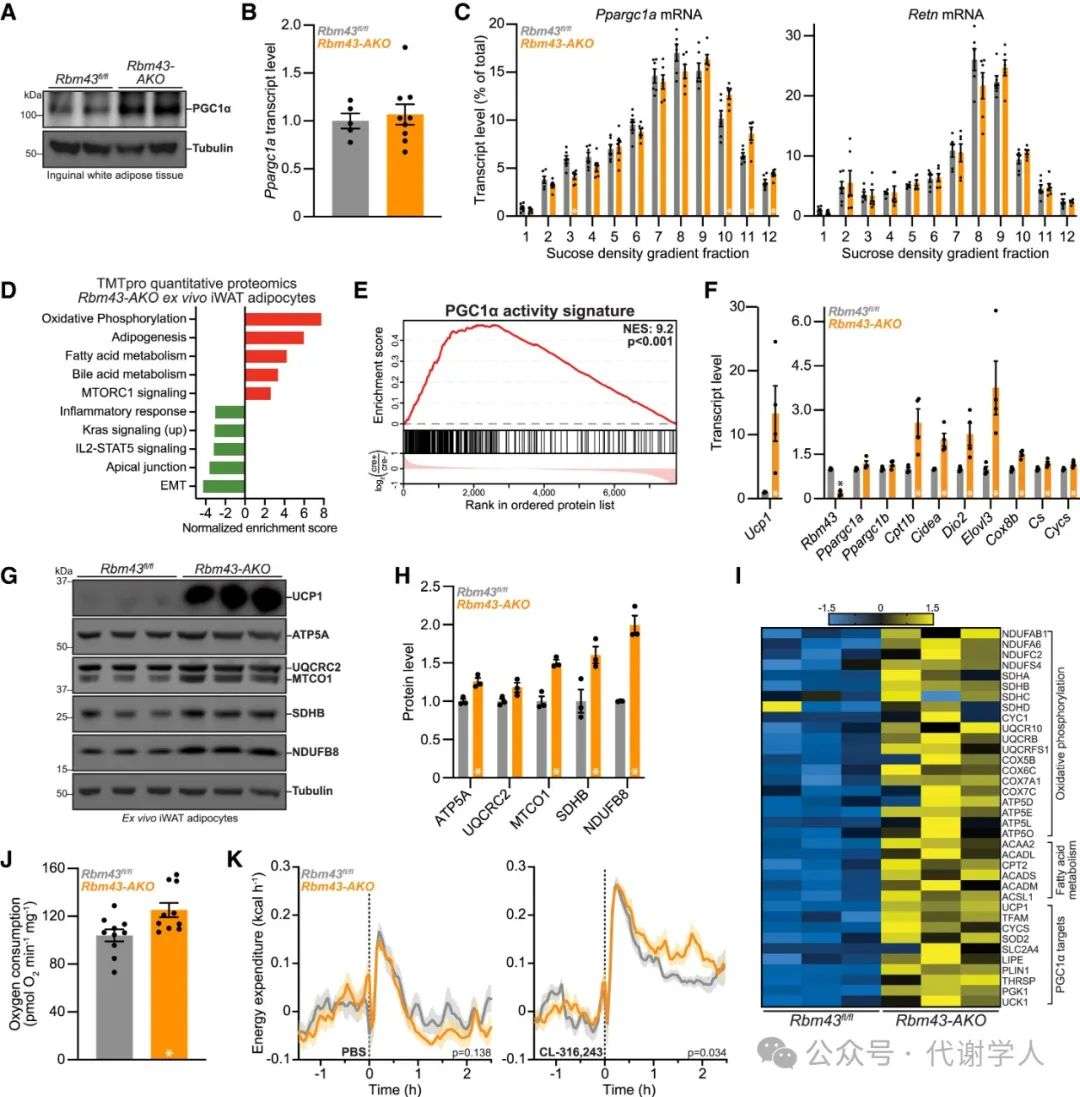
图4.RBM43调控白色脂肪中PGC1α的翻译水平、线粒体生物合成和氧化代谢
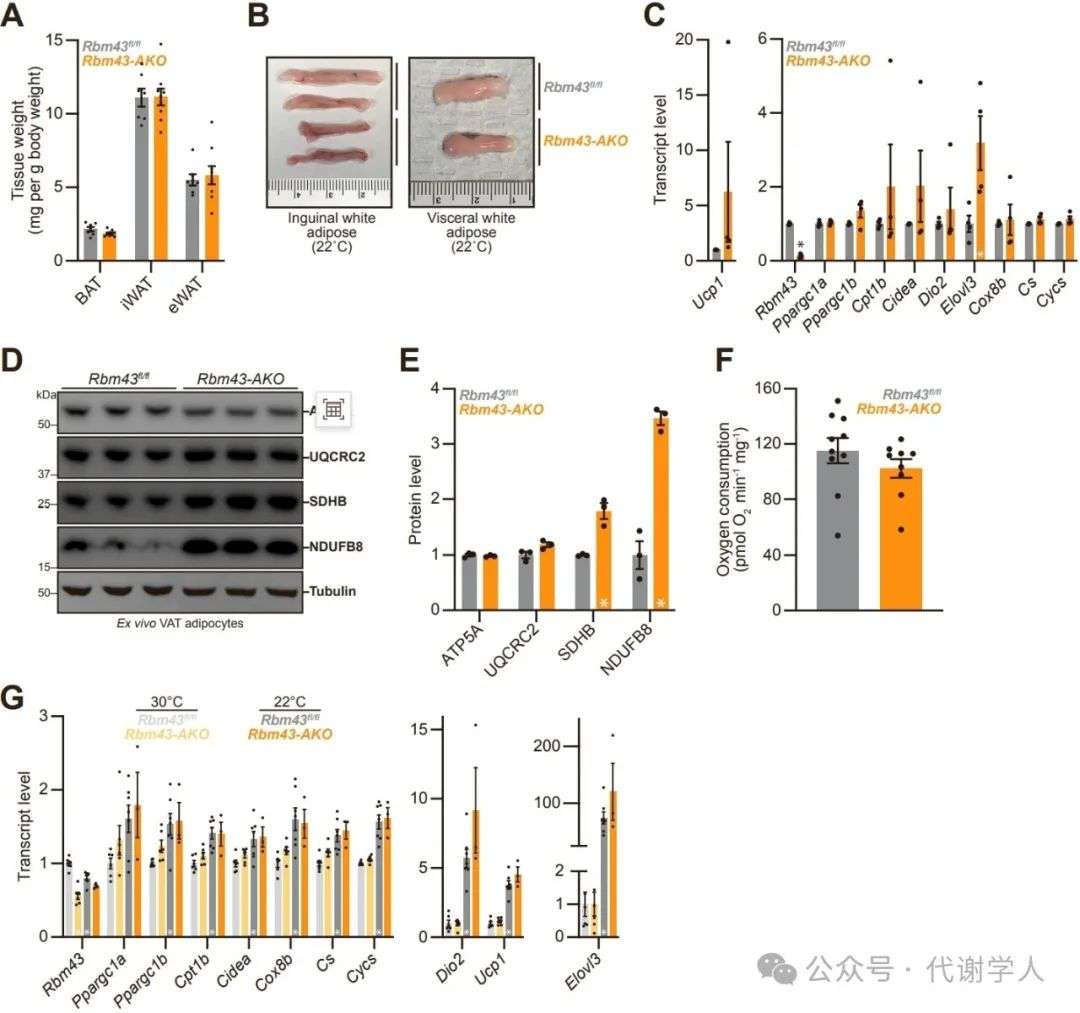
图S4.小鼠脂肪细胞中Rbm43特异性敲除
5.RBM43敲除可改善肥胖小鼠的糖耐量,并抑制脂肪炎症水平
由于Rbm43 AKO小鼠的脂肪产热能力提高,研究人员推测Rbm43 AKO小鼠或许可抵抗高脂饮食(HFD)诱导的肥胖。令人惊讶的是,在NCD或HFD喂养下,Rbm43 AKO小鼠的体重体脂与Rbm43fl/fl小鼠无显著差异(图5A、5B、5D和5E)。然而,HFD喂养下Rbm43 AKO小鼠的机体葡萄糖糖稳态明显改善,NCD喂养下Rbm43 AKO小鼠机体葡萄糖稳态无明显差异。这一现象在雌鼠和雄鼠中均可观察到(图5C、5F和S5)。
这些结果表明,脂肪Rbm43缺失可改善肥胖小鼠的代谢稳态,但这并不是由体重或机体能量消耗引起的。研究人员推测,Rbm43 AKO肥胖小鼠的代谢改善是否与脂肪炎症水平的变化有关,包括巨噬细胞趋化性或IFN信号。结果显示,Rbm43 AKO小鼠VAT中巨噬细胞含量下降(图5G和5H),这提示Rbm43可能通过调控WAT的炎症水平而非产热功能来影响肥胖小鼠的代谢稳态。
研究人员进一步探究了RBM43对VAT的调控作用。正如预期,Rbm43缺失显著上调了VAT脂肪细胞的PGC1α蛋白水平,对Ppargc1α基因表达无影响(图6A和6B)。随后,研究人员对Rbm43 AKO肥胖小鼠VAT脂肪细胞进行蛋白质组学分析,结果显示,Rbm43缺失后VAT脂肪细胞中PGC1α活性特征显著上调,包括氧化磷酸化复合物亚基基因和蛋白表达和OCR水平升高。这一现象在iWAT中也可观察到(图S6A-S6H)。通路富集分析显示,Rbm43 AKO小鼠VAT脂肪细胞中氧化磷酸化途径显著上调,值得注意的是,Rbm43缺失显著抑制了VAT脂肪细胞中NF-κB和IFN刺激的炎症信号以及EMT,这些途径被报道与肥胖密切相关(图6C-6D),且在Rbm43 AKO肥胖小鼠VAT脂肪细胞中整体蛋白质组呈现被抑制状态(图6E)。总之,这些结果表明,RBM43缺失抑制了脂肪中促炎和细胞外基质重塑程序的激活,进而改善肥胖相关代谢稳态。
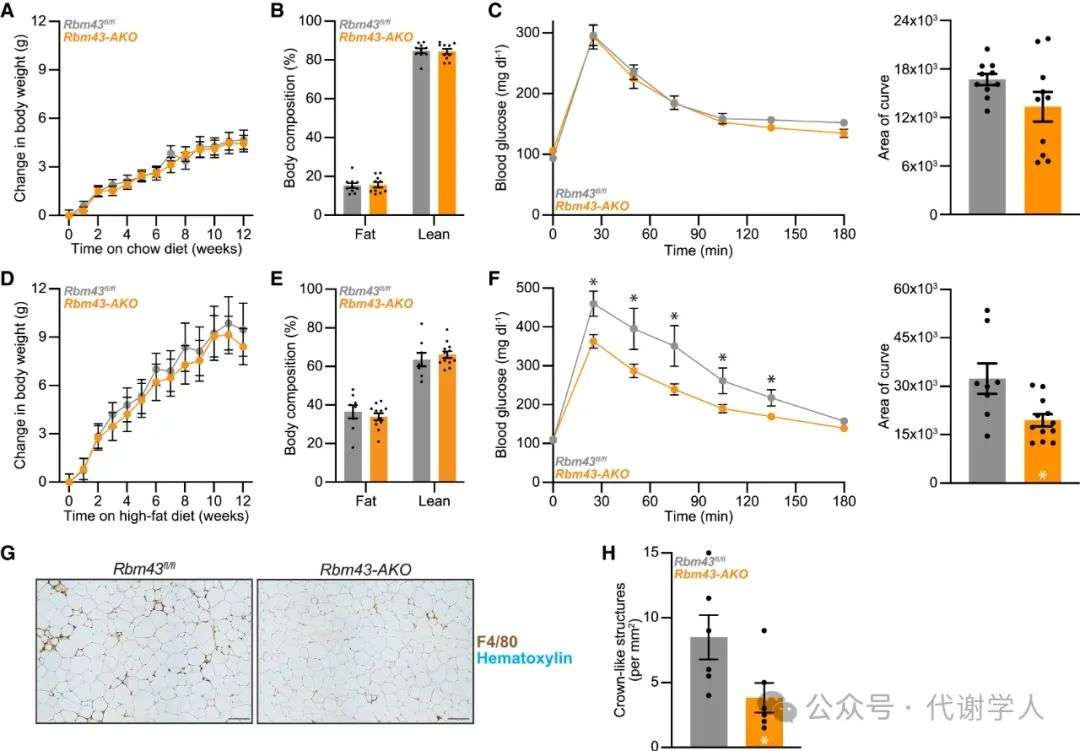
图5.脂肪细胞RBM43缺失改善了HFD小鼠的葡萄糖耐量,这与体重无关
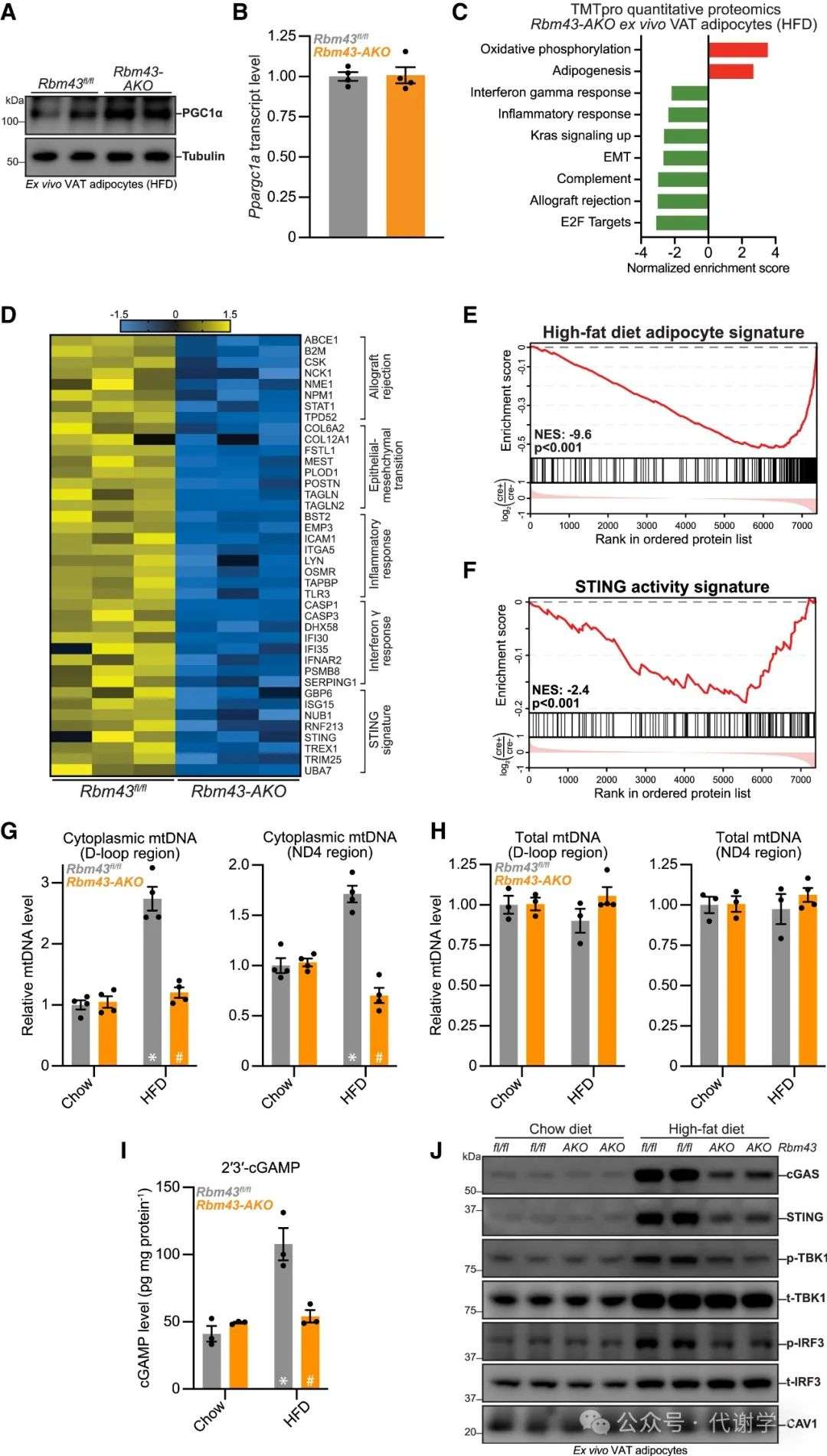
图6.RBM43促进肥胖小鼠白色脂肪细胞促炎基因表达和STING激活
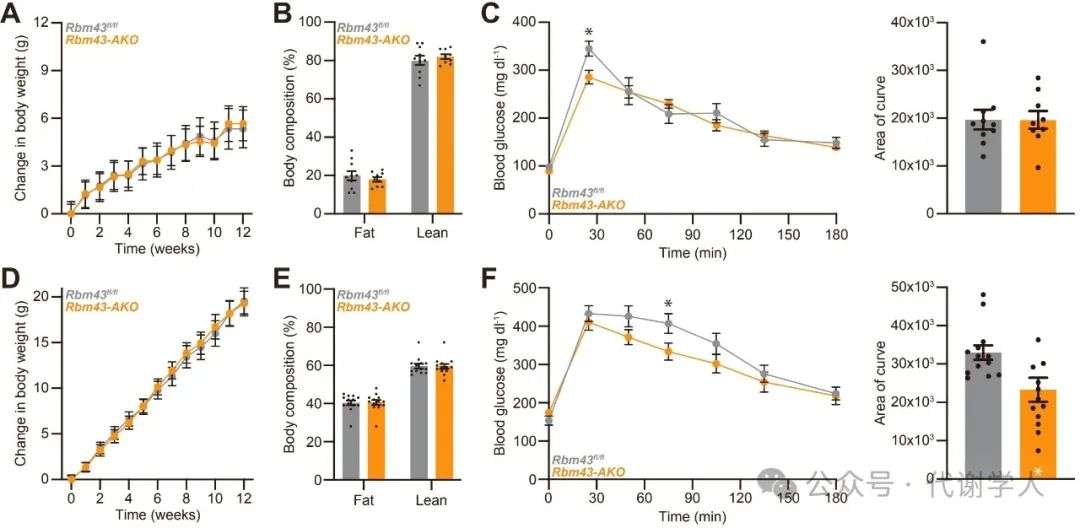
图S5.脂肪细胞RBM43缺失改善了HFD雄鼠的葡萄糖耐量,这与体重无关
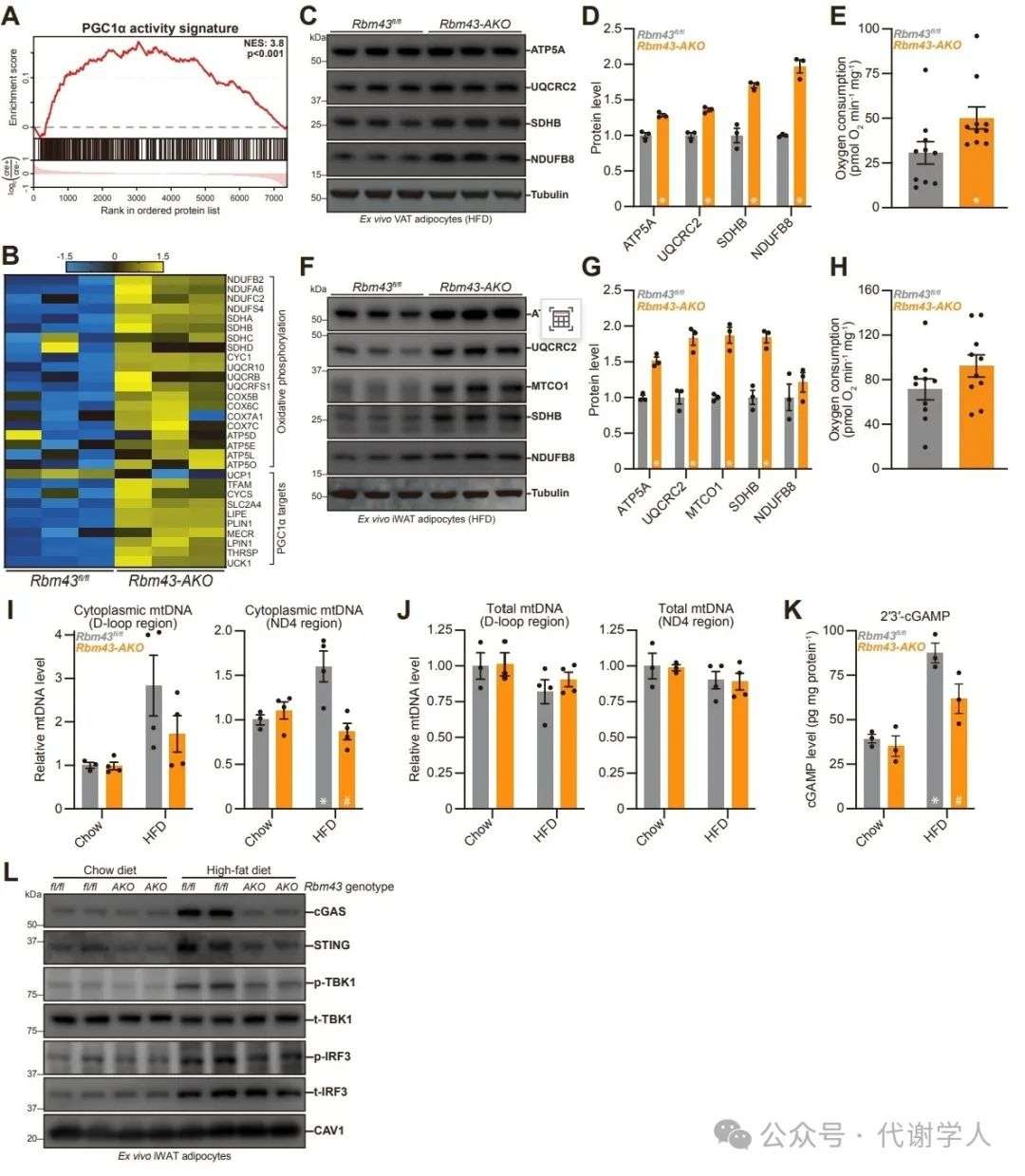
图S6.RBM43抑制肥胖小鼠白色脂肪细胞PGC1α并促进STING活化
6.RBM43促进胞质mtDNA积累和cGAS-STING激活,而PGC1α起保护作用
接下来,研究人员探究了RBM43是否通过调控上游信号途径来影响多种促炎基因表达程序。据报道,先天免疫传感器cGAS-STING信号可感应胞质DNA(包括病原体衍生DNA和定位异常的线粒体DNA(mtDNA)),并激活广泛炎症反应,且有研究报道mtDNA可促进肥胖小鼠脂肪组织STING信号。
因此,研究人员探究了RBM43对胞质mtDNA和STING信号的影响,结果显示HFD肥胖下Rbm43 AKO小鼠VAT脂肪细胞中胞质mtDNA水平显著减少,而细胞总mtDNA水平无显著变化,cGAS产物2,3-cGAMP水平下降,cGAS和STING蛋白水平下降,且cGAS-STING信号下游蛋白TBK1和IRF3的磷酸化水平下降,而NCD喂养下Rbm43 AKO小鼠VAT脂肪细胞中mtDNA水平与STING信号无显著差异(图6F-6J)。这一现象在iWAT和VAT脂肪细胞中均可观察到(图S6I-S6L)。这些结果表明,在肥胖状态下,脂肪RBM43缺失可保护脂肪细胞免受胞质mtDNA积累和STING信号激活的影响。
接下来,研究人员进一步探究了RBM43对PGC1α的抑制是否可促进肥胖状态下脂肪细胞中STING的激活。首先,研究人员检测了肥胖状态下Ppargc1α AKO小鼠(脂肪细胞特异性敲除Ppargc1α小鼠)VAT脂肪细胞中的cGAS-STING信号水平,结果显示Ppargc1α缺失显著提高了脂肪细胞胞质mtDNA水平,cGAS产物2,3-cGAMP水平以及cGAS-STING信号下游因子的蛋白表达和基因表达显著升高(图7A-7E)。值得注意的是,Ppargc1α AKO小鼠VAT脂肪细胞中Rbm43基因表达水平仅轻度增加(20%),这表明在肥胖状态下,PGC1α缺失和由此引起的STING激活并不引起脂肪细胞Rbm43表达的显著增加(即正反馈)(图S7A)。
随后,研究人员使用慢病毒CRISPR-Cas9靶向敲减iWAT原代脂肪细胞中Ppargc1α(图S7B),在Ppargc1α缺失的原代细胞中利用siRNA敲减Rbm43,Rbm43缺失对氧化代谢相关基因的表达无显著影响,这证实了Rbm43依赖于PGC1α来调控脂肪细胞的氧化代谢(图S7C)。Ppargc1α缺失显著提高了原代脂肪细胞中胞质mtDNA水平,尽管细胞总mtDNA水平下降。且2,3-cGAMP水平升高,cGAS-STING信号途径相关蛋白水平以及IFN诱导的基因表达水平上调(图7F-7J)。与此一致的是,对Ppargc1α-/-棕色脂肪细胞进行转录组分析,其上调基因主要富集在IFN反应中,并显著激活STING活性特征(图7K和7L)。使用STING小分子拮抗剂H151处理原代脂肪细胞可显著抑制由Ppargc1α缺失引起的IFN反应相关基因表达。相反,H-151不影响Rbm43介导的PGC1α抑制,表明STING活性不是RBM43功能所必需的(图7J、S7D和S7E)。这些结果表明PGC1α可抑制胞质mtDNA积累和cGAS-STING活性,这是RBM43抑制PGC1α改善肥胖相关代谢紊乱的潜在机制。
拓展阅读:PGC1α与mtDNA
PGC1α是线粒体生物发生和功能的关键调控蛋白,多数研究表明PGC1α可上调mtDNA的拷贝数。机制上,PGC1α可通过激活下游转录因子如NRF-1、NRF-2活性,促进TFAM(线粒体转录因子A)表达,进而促进mtDNA的转录和复制。线粒体膜通透性的改变是mtDNA释放到胞质的主要原因。PGC1α可通过增强线粒体氧化磷酸化能力,降低ROS水平,减少氧化应激对线粒体膜的损伤,进而抑制胞质mtDNA水平;此外,PGC1α还可通过Nrf2信号途径促进抗氧化酶如SOD2和GPx的表达,清除线粒体ROS,维持mtDNA的稳定性。
[1] Niranjana Natarajan, et al. Nat Commun. 2024. [2] Yao Zong, et al. Signal Transduct Target Ther. 2024. [3] Phillip A Dumesic, et al. Cell Metab. 2019. [4] Ji-Hye Song, et al. Phytomedicine. 2024.
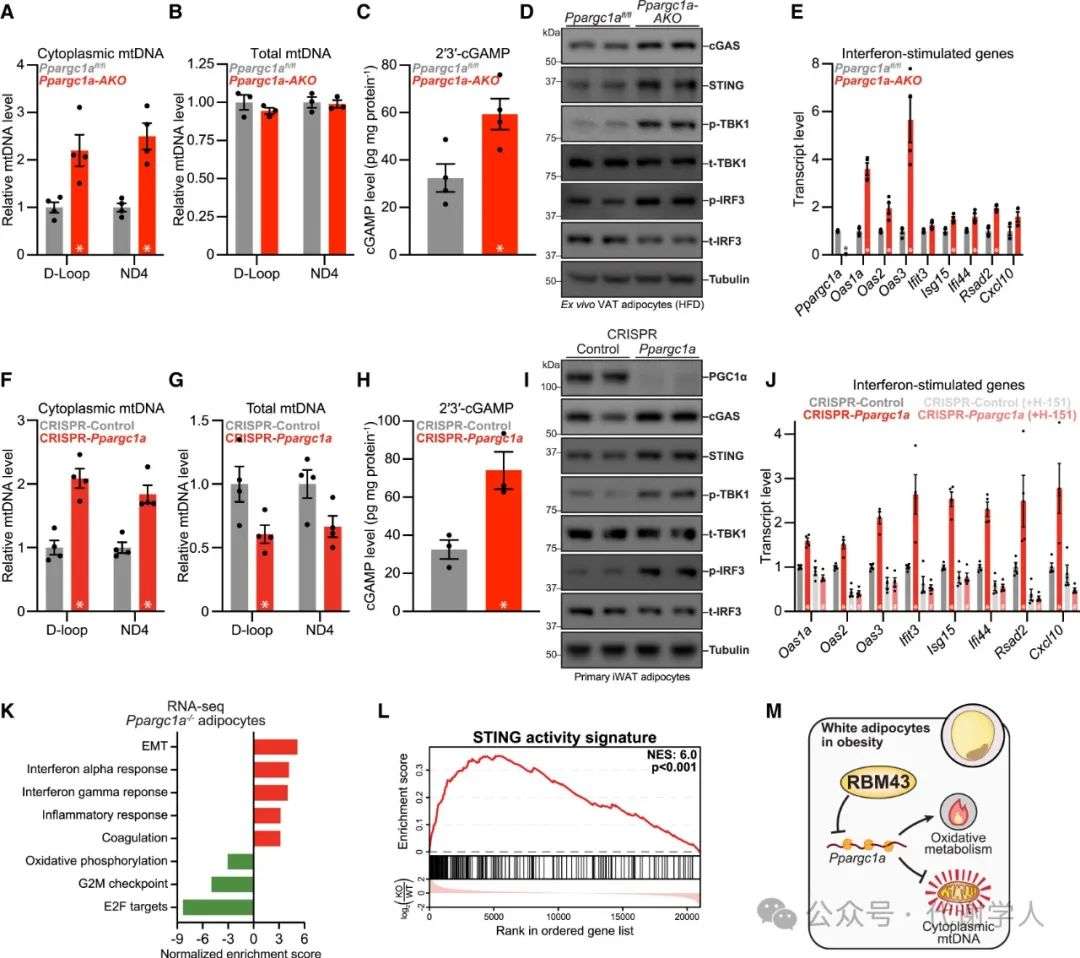
图7.PGC1α抑制细胞质mtDNA积累和STING激活
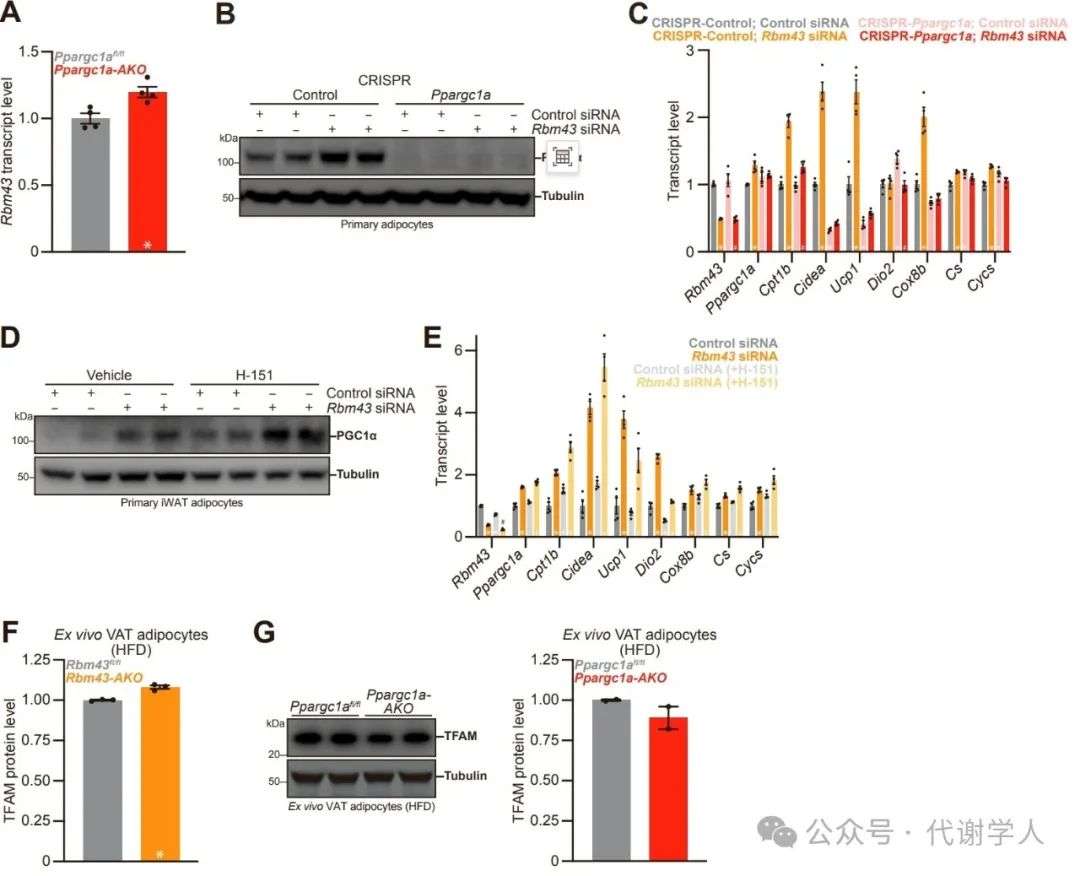
图S7.RBM43对氧化代谢基因表达的调控依赖于PGC1α,而不依赖于STING活性
总结:
本文中,研究人员发现炎症信号能诱导RBM43表达上调,这会显著抑制PGC1α的翻译,从而抑制脂肪细胞的线粒体生物合成和氧化代谢。在肥胖状态下,敲除RBM43可以改善葡萄糖稳态,减少脂肪组织炎症,并抑制脂肪细胞中cGAS-STING信号通路。研究人员进一步发现,PGC1α能抑制细胞质中mtDNA积累和cGAS-STING信号通路激活。总之,这些发现为研究肥胖相关代谢疾病的发病机制提供了新的视角,并能为开发新的治疗策略提供理论基础。
链接:https://www.sciencedirect.com/science/article/pii/S1550413125000130?via%3Dihub
https://wap.sciencenet.cn/blog-3483272-1483236.html
上一篇:Cell Metabolism:美食诱惑,神经”上火”
下一篇:Cell Metabolism:“肌”不可失——线粒体的“钙”世英雄