博文
Phenomics | 复旦大学郑琰团队研究揭示DNA提取方法对肠道微生物组分析的影响
|
近日,《表型组学(英文)》(Phenomics)在线发表了复旦大学生命科学学院/人类表型组研究院郑琰研究员团队题为“Impact of DNA Extraction Methods on Gut Microbiome Profiles: A Comparative Metagenomic Study”的研究论文。该研究系统评估了八种常用的DNA提取方法对肠道微生物组分析的影响,揭示了不同提取方法在微生物组组成、多样性和功能分析中的显著差异,为肠道微生物组研究的标准化和可靠性提供了重要依据。
论文DOI链接:
https://doi.org/10.1007/s43657-025-00232-x
论文引用格式:
Pu, Y., Zhou, X., Cai, H. et al. Impact of DNA Extraction Methods on Gut Microbiome Profiles: A Comparative Metagenomic Study. Phenomics (2025). https://doi.org/10.1007/s43657-025-00232-x
研究背景
肠道微生物组研究依赖于DNA提取技术的准确性,但不同方法可能导致实验结果偏差,甚至掩盖真实的生物学信号。既往研究(如MetaHIT、HMP计划)多依赖非标准化或已停产的提取方案,且缺乏对真菌、古菌等多界微生物的系统评估。随着自动化技术与跨物种(trans-kingdom)研究需求的增长,亟需对新型商业化试剂盒的性能进行系统性验证,以保障数据可靠性、研究可重复性及跨研究可比性。
研究方法
研究团队采用模拟微生物群落(MMC,含14种细菌/1种古菌/2种真菌)和添加外源微生物的人类粪便样本,评估了八种商业DNA提取试剂盒的性能,包括Qiagen、Tiangen和ZymoBIOMICS等主流品牌。通过鸟枪法宏基因组测序,系统分析各方法在DNA产量、纯度、微生物群落结构(细菌、古菌、真菌)及功能通路恢复能力上的差异。研究引入五维评价体系(灵敏度、假阳性率、多样性、相似性、技术重复性),并结合自动化流程和成本效益分析,综合筛选最优方案。
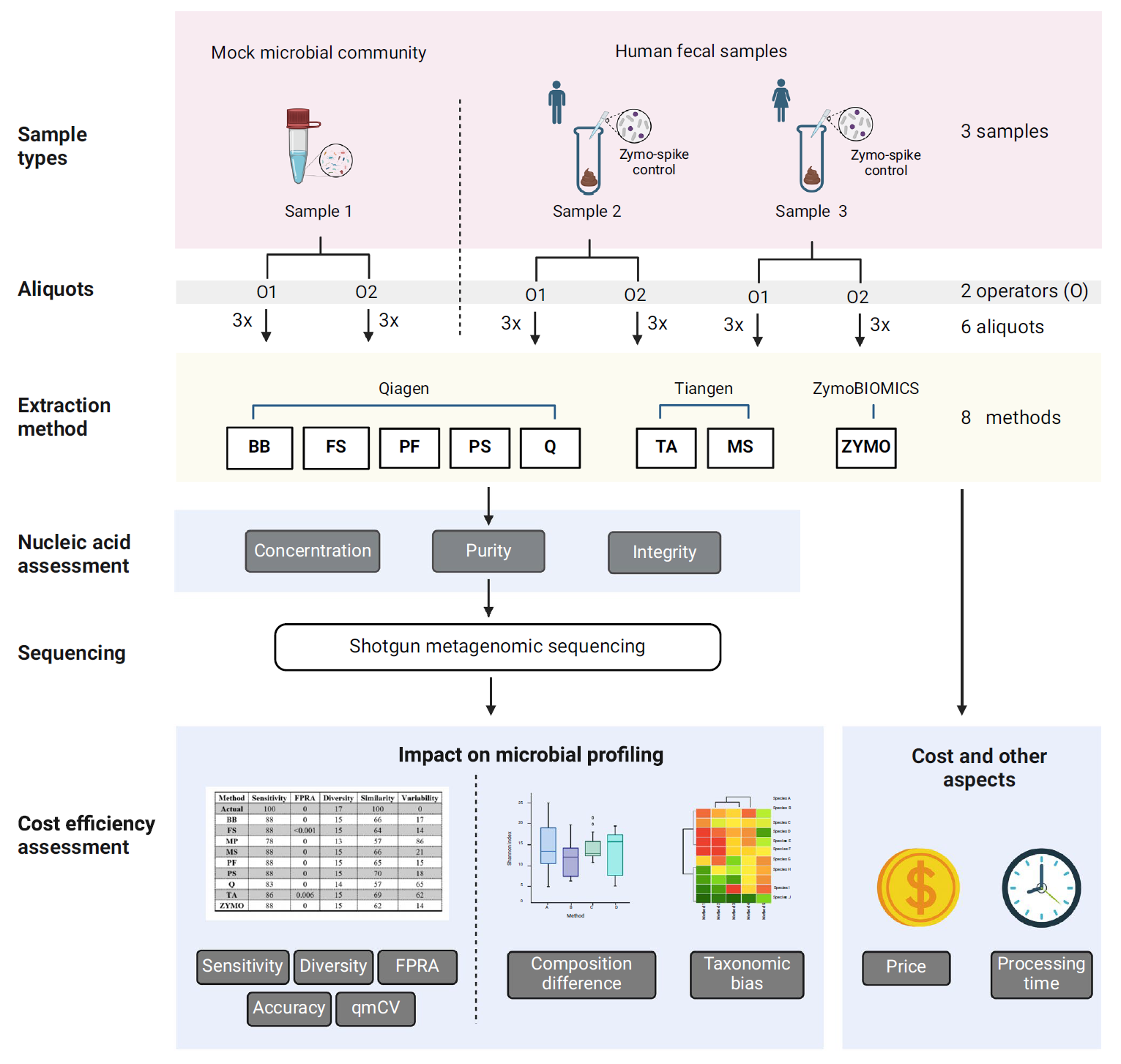
图 1 研究设计流程图
研究结果
研究结果显示,所有方法均能提供足够的DNA进行宏基因组测序,但不同方法在微生物群落重建准确性方面存在显著差异。使用模拟微生物群落(MMC)时,QIAamp PowerFecal Pro Kit(PF)和DNeasy PowerSoil HTP kit(PS)展现出与理论组成最高相似性和技术重复最低变异性。在真实粪便样本中,提取方法贡献了21.4%的微生物组变异,显著影响32%检测物种的丰度,其中89%为革兰氏阳性菌。采用小粒径的研磨珠进行机械裂解的PF和PS方法对厚壁菌门等革兰氏阳性菌的提取效率更高,能捕获更丰富的微生物多样性,但成本相对较高。
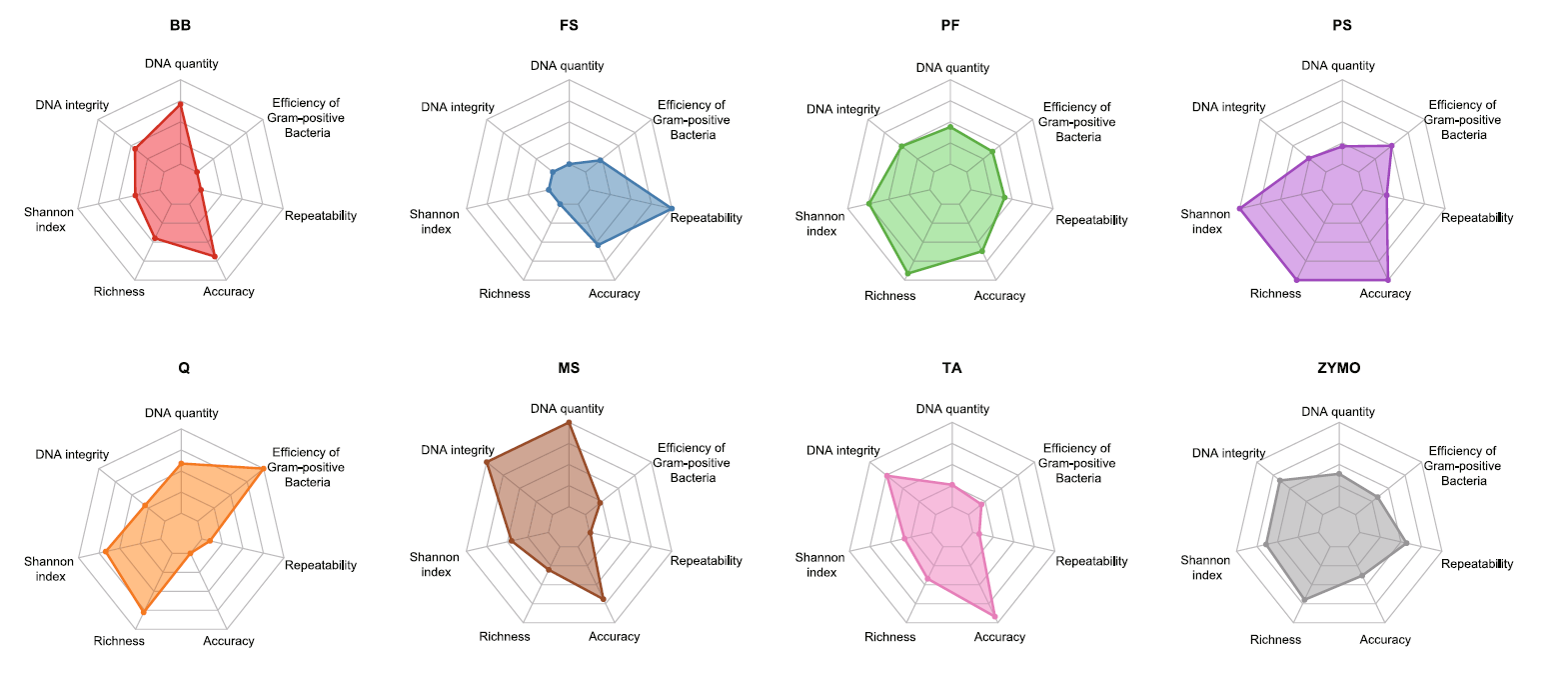
图 2. 不同DNA提取方法的性能比较
研究结论
本研究评估了八种广泛应用DNA提取方法的效能,推荐PS(DNeasy PowerSoil HTP)与PF(QIAamp PowerFecal Pro)方法作为肠道微生物组研究的优选方案,因其在微生物组成分析中偏差最小,且在时间与人力成本上具有更高效率。研究结果进一步强调,采用一致且可靠的DNA提取方法对提升肠道微生物组研究的可重复性至关重要,同时凸显在整合基于不同方法获得的数据时,需将DNA提取技术作为关键技术因素纳入考量,以减少技术偏倚对科学结论的干扰。
研究意义
该研究为肠道微生物组的DNA提取方法提供了系统的对比数据,为今后的微生物组研究提供了重要的技术参考。通过优化DNA提取方法,研究人员能够更加精确地分析肠道微生物群落,进一步推动微生物学与人体健康研究的深入发展。
复旦大学生命科学学院/人类表型组研究院郑琰研究员为通讯作者,蒲彦霓博士后和周小锋博士后为第一作者。该项目获得国家重点研发计划(2021YFA1301000)和和上海市科技重大项目(2023SHZDZX02)的资助。
Abstract
In gut microbial research, DNA extraction remarkably influences study outcomes and biological interpretations. Rapid advancements in the research scale and technological upgrades necessitate evaluating new methods to ensure reliability and precision in microbial community profiling. We systematically evaluated the performance of eight recent and commonly used extraction methods using a microbial mock community (MMC) and fecal samples from two healthy volunteers, incorporating bacterial, archaeal, and fungal constituents. Performance metrics included nucleic acid assessment, microbial profile assessment,
and scalability for large-scale studies, leveraging shotgun metagenomics for in-depth analysis. Despite variations in
DNA quantity and quality, all methods yielded sufficient DNA for shotgun metagenomic sequencing. In the MMC microbial profile assessment, the QIAamp PowerFecal pro Kit (PF) and DNeasy PowerSoil HTP kit (PS) methods exhibited higher similarity with the theoretical composition and lower variability across technical replicates compared to other methods. For fecal samples, the extraction method accounted for 21.4% of the overall microbiome variation and significantly affected the abundances of 32% of detected microbial species. Methods using mechanical lysis with small beads, such as PF and PS, demonstrated better efficiency, indicated by increased microbial diversity in extracting DNA from Gram-positive bacteria. Furthermore, the PF and PS methods are notably simple to execute and automation-friendly, though relatively costly. Our study underscores the importance of maintaining consistency in DNA extraction methods for reliable comparative metagenomic analyses. We recommend PF and PS methods as optimal for expansive gut metagenomic research, emphasizing the critical role of mechanical lysis in DNA extraction.
作者简介
通讯作者

郑琰,现任复旦大学生命科学学院研究员,博士生导师。国家“万人计划”青年拔尖人才,上海高校特聘教授(东方学者),入选“全球前2%顶尖科学家榜单”(2023、2024)。近年来,共计主持人才项目6项和科研项目4项(国家自然科学基金2项,国家重点研发计划2项)。担任Molecular Genetics and Genomics杂志副主编,先后发表SCI文章100余篇,其中以重要作者身份在JAMA、Nature Reviews Endocrinology、BMJ、Diabetes Care、Natl Sci Rev等杂志上发表文章67篇。连续多年被评为中国高被引学者并受到国内外媒体报道。主要从事肥胖相关代谢病的人群流行病学因素,尤其是利用代谢组及微生物组学大数据,发现肥胖相关疾病的新型生物标志物,探索饮食影响健康的代谢中介机制。
第一作者

蒲彦霓,复旦大学人类表型组研究院博士后。主要从事增龄性疾病的分子流行病学研究。曾获得上海市“超级博士后”计划和复旦大学“超级博士后”计划资助。近年来,以第一作者(含共同)在Nature Aging,Phenomics,mSphere发表SCI论文。

周小锋,复旦大学人类表型组研究院博士后。研究方向为肠道菌群对代谢性疾病的影响,主要通过流行病学、统计学以及生物信息学方法探索肠道菌群在代谢性疾病发生发展过程中的作用。
https://wap.sciencenet.cn/blog-3558836-1486710.html
上一篇:Phenomics | 上海儿童医学中心王纪文/王英燕团队与复旦大学王炜研究员揭示生酮饮食下肠道细菌的在体生长动态变化
下一篇:Phenomics | 兰州大学第一医院齐平团队发表揭示肠道稳态与胰腺癌潜在关联的综述,为治疗提供新视角
全部作者的其他最新博文
- • Phenomics | 中山大学肿瘤防治中心曾木圣院士团队发表综述:肿瘤诊疗新靶点,整合素α6靶向技术引领精准医疗新时代
- • Phenomics | 上海交通大学医学院张孝勇教授团队开发磁共振成像降噪新方法,有潜力改善脑小血管病的诊断效能
- • Phenomics | 复旦大学倪挺教授团队揭示基因内含子多聚腺苷酸化调控细胞衰老新机制
- • Phenomics | 加拿大阿尔伯塔大学和复旦大学联合开发基于文本、音频和视频的多模态抑郁症检测与评估方法
- • Phenomics | 翟振国/蒋太交/张鹏团队合作开发自适应多基因风险评估模型提升汉族人群静脉血栓栓塞症的风险预测能力
- • 祝贺 | 《表型组学(英文)》成功入选2025年“中国科技核心期刊”