博文
Materials Studio | 新能源车续航2000km不是梦 Nat.Commun重磅发布 高能量密度锂硫电池
|

【趣事分享】
该文献中采用Ca²⁺添加剂的锂硫软包电池性能优异:E/S=2.4μLmg-¹时能量密度493Whkg-¹,220次循环容量保持率70%;锂硫软包电池的能量密度493Whkg-¹(0.493KWhkg-¹)经过换算,可以计算出200KWh的锂硫软包电池重400Kg;锂硫软包电池不仅重量轻,而且200度电池可以续航接近2000km。
【文献介绍】
近日,韩国科学技术院化学及生物分子工程系金熙德教授团队在《Nature Communications》期刊上发表了关于稀薄电解质锂硫电池的可转换和再生路易斯酸性电解质添加剂的文章。该文献聚焦于锂硫电池中路易斯酸性Ca²⁺添加剂的应用,其中Materials Studio模拟理论计算(DFT和MD计算)从原子和分子层面深入剖析了添加剂作用机制,为实验现象提供理论支撑,对理解电池性能提升原理至关重要。
要实现具有高能量密度的实用化锂硫电池,贫电解液设计是关键。然而,在低电解液/硫(E/S)比条件下,电解液相中高浓度的多硫化锂会对电池的循环性能和容量产生限制。研究提出添加少量路易斯酸性钙离子(Ca²⁺)解决该问题——Ca²⁺可将多硫化锂转化为硫化钙(CaS)与硫单质(S₈),避免电解液堵塞、多硫化物穿梭及锂腐蚀,原位生成的CaS还能催化多硫化锂还原,且充电时CaS氧化再生Ca²⁺,实现“Ca²-CaS”循环长效作用。采用该添加剂的锂硫软包电池性能优异:E/S=2.4μLmg-¹时能量密度493Whkg-¹,220次循环容量保持率70%;E/S=2.9μLmg-¹时能量密度346Whkg-¹,360次循环保持率77%。该研究融合锂硫与钙硫化学,为突破电池“能量密度-循环稳定性”难题提供有效方法,推动贫电解液锂硫电池实用化。

【研究背景】
锂硫电池因高理论能量密度等优势极具潜力,但多硫化物(LiPSs)穿梭、硫还原反应(SRR)动力学迟缓及锂金属阳极(LMA)腐蚀等问题阻碍其发展。降低电解质/硫(E/S)比虽可提升能量密度,却会加剧上述问题,开发有效电解质添加剂迫在眉睫。
【理论研究方法】
1、密度泛函理论(DFT)计算:运用Materials Studio中的DMol3模块,采用广义梯度近似(GGA)的Perdew-Burke-Ernzerhof(PBE)泛函和双数值加极化(DNP+)基组,对分子几何结构进行优化并计算结合能。设置轨道截断值为4.0Å,利用COSMO模型模拟DME溶剂环境,以0.05eV的高斯展宽计算硫化钙和石墨烯与LiPSs的结合能及相对自由能,在催化剂最暴露的(220)表面进行计算。
2、分子动力学(MD)模拟:借助Materials Studio的Forcite模块构建模拟电池体系,设定体系中包含1.6MLi₂S₄(对应E/S比为5μLmg⁻¹、硫负载为3.8mgcm⁻²时硫全部溶解的浓度),采用COMPASSIII力场描述分子间相互作用。在1atm、298K条件下,先在NPT系综平衡1ns用于Li⁺配位分析,再经1ns NVT系综模拟后进行5ns模拟,通过径向分布函数获取分子间配位化学信息。
【结果和讨论】
1. CaS对LiPSs的吸附及催化作用
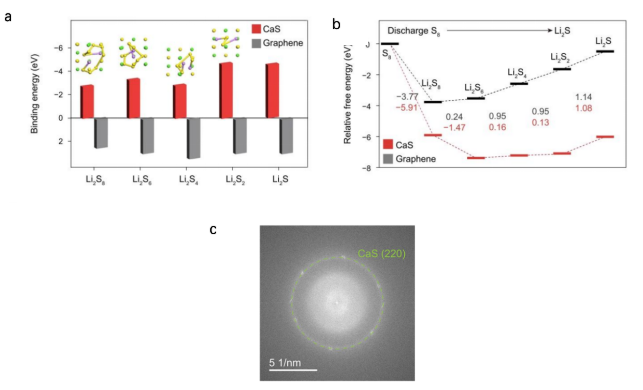
图1.a密度泛函理论(DFT)计算相关图示:黄色、绿色和紫色球体分别代表硫(S)、钙(Ca)和锂(Li)原子;插图展示多硫化锂(LiPSs)在CaS表面吸附的优化构型。bDFT计算得到的S₈在CaS表面和石墨烯表面放电生成Li₂S过程的自由能图。C放电至2.1V的硫正极选区电子衍射(SAED)图谱
为探究硫化钙(CaS)的催化作用,文献采用密度泛函理论(DFT)计算了不同多硫化物(PS)物种在石墨烯表面与CaS表面的结合能。计算过程中,CaS表面基于透射电子显微镜(TEM)分析(图1c)所确定的原位生成CaS的(220)晶面构建。图1a对比了不同链长的多硫化锂(Li₂Sₙ,n=8、6、4、2、1)在CaS表面与石墨烯表面的结合强度:石墨烯表面仅存在弱范德华相互作用,缺乏强吸引力;而CaS表面则通过其硫位点与锂离子(Li⁺)产生静电相互作用。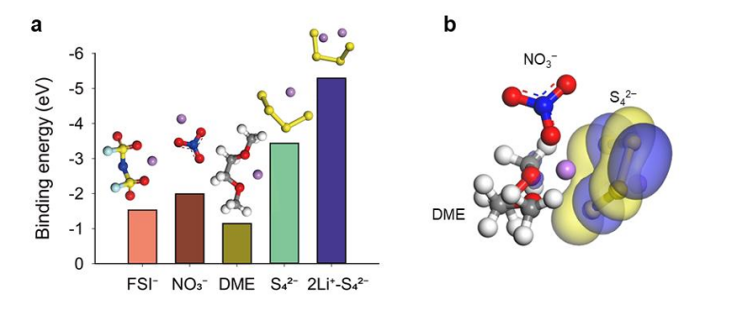
图2.(a)采用密度泛函理论(DFT)计算得出的氟磺酰亚胺根(FSI-)、硝酸根(NO3-)、四硫离子(S42-)和硫代硫酸根(LiS4-)与锂离子(Li+)的结合能。(b)对照电解液中锂离子(Li+)的溶剂化结构。在电解液组分中,多硫化物(PS,即S42-)与锂离子(Li⁺)的结合能最高。对于包含S42-的锂离子溶剂化物,最低未占据分子轨道(LUMO)分布在S42-周围。
钙基电解液对多硫化物(PS)溶解能力的改变影响着锂金属的腐蚀情况和稳定性。在对照电解液中,像S₄²⁻这样的短链多硫化物倾向于与锂离子(Li⁺)紧密结合
2. Ca²⁺对Li⁺溶剂化结构的影响
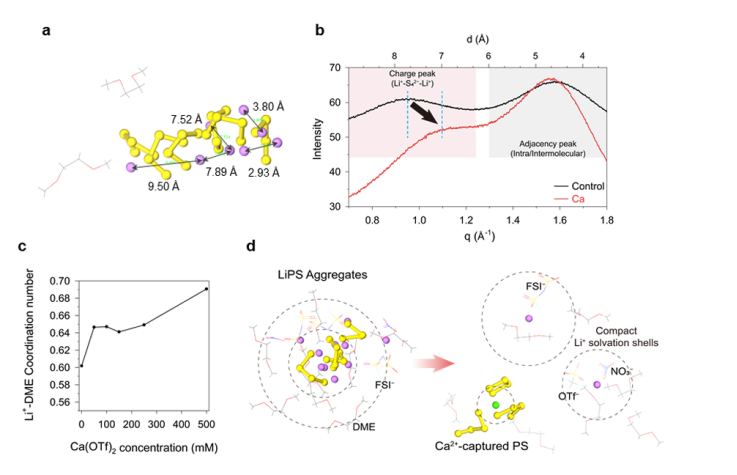
图3.(a)对照正极电解液中多硫化锂(LiPS)聚集体的分子动力学(MD)模拟箱快照。锂(Li)、硫(S)、碳(C)、氧(O)原子分别用紫色、黄色、灰色和红色的球或棍状模型表示。(b)对照电解液与含钙(Ca)电解液的小角X射线散射(SAXS)图谱,两种电解液中多硫化锂浓度均为1.6mol/L(Li₂S₄),与分子动力学模拟所用浓度一致。长度d根据布拉格定律(公式:d=2π/q)计算得出。(c)通过分子动力学模拟得到的电解液中,与锂离子(Li⁺)配位的1,2-二甲氧基乙烷(DME)分子平均数量。(d)对照正极电解液(左)与0.05mol/L含钙正极电解液(右)中,锂离子(Li⁺)、钙离子(Ca²⁺)及四硫离子(S₄²⁻)周围分子分布的分子动力学快照。氮(N)原子用蓝色棍状模型表示。
为阐明多硫化物(PS)溶解度降低的背后机制,开展了分子动力学(MD)模拟。通过MD模拟研究Ca²⁺添加剂存在时Li⁺溶剂化壳层结构变化,分析Li⁺与S₄²⁻、FSI⁻阴离子等的配位情况,以及这些变化对电解液性质和固体电解质界面(SEI)形成的影响。Li⁺溶剂化结构变化:MD模拟显示,添加Ca²⁺后,Li⁺与S₄²⁻的平均配位数从1.73降至1.05,与FSI⁻阴离子的配位数从0.26升至0.46,Li⁺溶剂化鞘层中阴离子/PS比例从0.15/1.0提高到0.44/1.0。这表明Ca²⁺添加剂改变了Li⁺溶剂化结构,增强了Li⁺-FSI⁻配位,减少了LiPSs的溶解,为形成富LiF的SEI提供了条件。
【总结与展望】
理论计算并非独立存在,而是与实验形成“假设-验证-解释”闭环:通过DFT明确CaS的催化本质,通过MD揭示Li⁺溶剂化调控机制,二者共同支撑“Ca²⁺-CaS互转化”的核心设计,为实验中电池高能量密度、长循环稳定性提供原子层面的合理解释,也为后续多价阳离子添加剂设计提供理论参考。
【文章信息】
原文标题:InterconvertibleandrejuvenatedLewisacidicelectrolyteadditiveforleanelectrolytelithiumsulfurbatteries
标题中文翻译:用于贫电解液锂硫电池的可互转化且可再生的路易斯酸性电解液添加剂
期刊:《naturecommunications》
DOI:https://doi.org/10.1038/s41467-025-62169-z
通讯作者单位:韩国科学技术院;化学及生物分子工程学系
End

https://wap.sciencenet.cn/blog-3536821-1519265.html
上一篇:【直播预告】东方科软×月只蓝:直击Materials Studio痛点、难点,10+热门问题,让MS模拟更加顺畅!
下一篇:Discovery Studio文献解读 | 结核杆菌FtsZ抑制剂计算药理学研究