博文
Materials Studio文献解读 | 重庆大学 | Al-5Ti-B细化剂为何在Al-Si合金中失效?
|

【文献介绍】
本研究由重庆大学李谦教授团队联合通用汽车中国科学实验室等单位在《Acta Materialia》上发表,系统研究了Al–Si铸造合金中使用Al–5Ti–B晶粒细化剂时出现的“Si中毒”现象。通过结合高分辨率扫描透射电子显微镜(STEM)、第一性原理计算(CASTEP) 与热力学计算(CALPHAD),揭示了Si中毒的微观机制,并推翻了长期以来“硅化物覆盖TiB₂导致中毒”的传统观点。研究指出,Si原子在TiB₂/α-Al界面处的TiAl₃二维化合物中偏聚,并通过形成强Ti–Si共价键,破坏了TiAl₃ 2DC的结构与化学环境,从而阻碍α-Al的异质形核。
本研究通过CASTEP模块进行的第一性原理计算,揭示Si中毒原子级机理。其主要完成以下的核心工作:
(1)构建并优化了关键的界面原子模型:构建了TiB₂/TiAl₃ 2DC(二维化合物)和TiB₂/TiAl₃ 2DC/α-Al的系列超胞模型。该模型涵盖了界面与不同Si掺杂浓度(8.3 at.%和20.8 at.%)的界面,准确模拟了实验观察到的Si偏聚情况。
(2)量化了Si对界面结构有序度的破坏:通过计算优化后模型的原子对分布函数(PDF),定量分析了TiAl₃ 2DC和其上方外延α-Al层的结构有序度。结果表明,Si的掺入导致PDF特征峰显著宽化和分裂,从理论上证明了Si会引起TiAl₃ 2DC和α-Al的晶格畸变与无序化。
(3)揭示了界面电子结构与化学键合的演变:通过电荷密度差分图,直观展示了清洁界面处Ti-Ti和Ti-Al原子间存在强烈的电荷积累和电子云重叠,形成了强键合。通过电荷密度分析,揭示了Si掺杂后,电荷在Ti-Si原子间重新聚集,形成了强的Ti-Si共价键。
CASTEP的计算结果表明,通过上述核心工作,CASTEP计算从 “原子结构”→“能量”→“电子性质” 三个维度,完整地揭示了Si中毒的微观机理:结构上,Si引起模板和晶核的晶格无序。能量上,Si降低模板稳定性并提高形核能垒。化学上,Si通过形成强Ti-Si键,竞争性地削弱了界面处关键的Ti-Ti和Ti-Al键。这些计算结果为 “Si偏聚于TiAl₃ 2DC导致中毒” 的新机制提供了坚实的理论证据,彻底颠覆了传统的“硅化物涂层”假说。
本研究基于CASTEP模块的第一性原理计算,对铝合金铸造过程中容易产生“Si中毒”这一经典问题给出了全新的机理解释。研究团队在实验中并未发现传统理论所预测的块状硅化物涂层,而是通过原子尺度的成分分析,首次清晰地观察到有5-20 at.%的Si原子偏聚于TiB₂与α-Al之间的关键中间层——TiAl₃二维化合物(2DC)中。随后的理论计算表明,这些偏聚的Si会与Ti形成强共价键,这不仅扰乱了TiAl₃ 2DC本身及其上方外延α-Al的晶体结构有序性,还显著降低了该中间层的热力学稳定性及其与α-Al之间的化学相互作用,最终从结构、能量和电子三个层面共同阻碍了α-Al的有效形核。

【研究背景】
Al–5Ti–B是铝铸合金中最常用的晶粒细化剂,但在含Si的铸造合金(如Al–Si系)中其细化效果显著下降,此现象称为“Si中毒”。过去60年间,学界普遍认为Si会促使硅化物(如TiSi₂、Ti₅Si₃等)在TiB₂表面析出,阻碍α-Al形核。然而,该机制缺乏直接实验证据,且无法解释所有实验现象。本研究从界面原子结构与电子结构出发,重新审视Si中毒的本质。
【理论研究方法】
本研究基于CASTEP模块的第一性原理计算,将“Si中毒”的根源从宏观的相覆盖重新定位到原子尺度的界面偏聚与键合重构,为设计抗中毒的新型晶粒细化剂提供了清晰的理论基础和研究方向。
相关参数设置:

配对分布函数(Pair Distribution Function,PDF):是一种描述材料中原子是如何在空间中排布的“尺子”,它通过统计不同距离上的原子对数量,来揭示材料的原子级结构信息,无论是规则排列的晶体,还是无序排列的非晶体。
【结果和讨论】
1.晶体结构模型
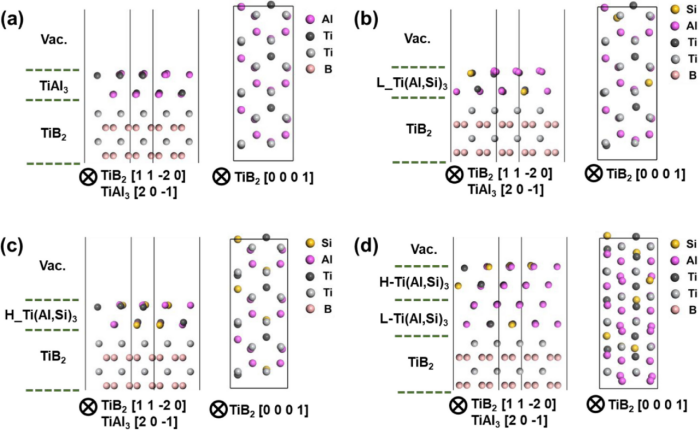
图1.含有TiAl3的超胞的弛豫结构2DC
(a)TiB2|TiAl3|真空;(b)TiB2|L_Ti(Al,Si03|真空;(c)TiB2|H_Ti(Al,Si)3|真空;(d)TiB2|L_H_Ti(Al,Si)3|真空。
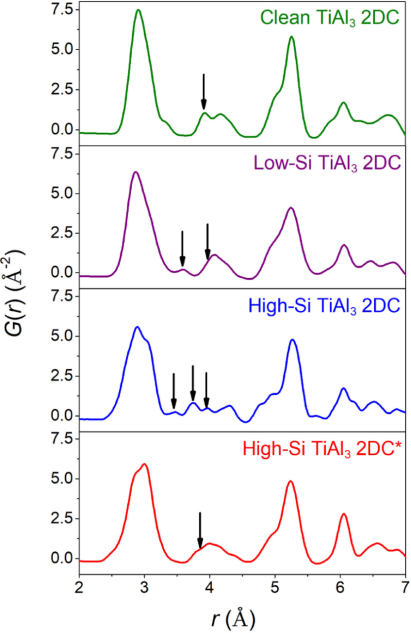
图2.弛豫TiAl的原子PDF计算结果32DC
图1和图2通过DFT计算从理论上验证了Si的破坏作用。弛豫后的结构模型显示Si掺杂导致原子排列畸变。对应的原子对分布函数图表明,特征峰发生宽化和分裂。这定量地证明了Si的引入会显著破坏TiAl₃ 2DC的原子级长程有序。
2.纯TiB2与Si掺杂TiB₂/TiAl₃界面处的差分电荷密度
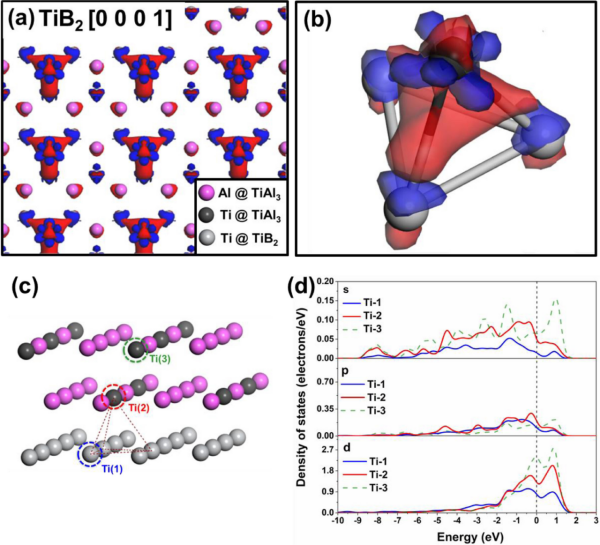
图3(a)纯TiB/TiAl3界面处的电荷密度差异(红色:积累;蓝色:耗尽);(b)电子积累区的特写视图;(c)PDOS所考虑的Ti原子的位置;(d)PDOS光谱。
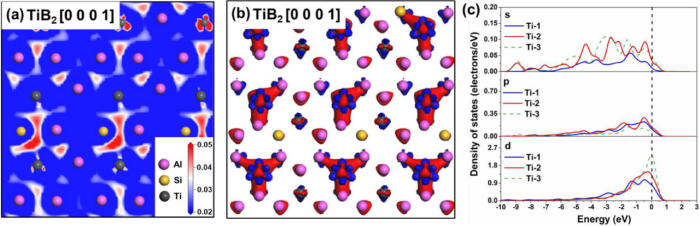
图4
(a)低硅掺杂TiB2/TiAl3 2DC界面处TiAl3(112)晶面的上的电荷密度变化;(b)低硅掺杂TiB2/TiAl3 2DC界面处的电荷密度差异,等值面值为±0.05 e/Å3(红色:积累;蓝色:耗尽);(c)低硅掺杂TiB2/TiAl3 2DC界面周围的Ti原子的PDOS。Ti的位置与图3(c)中的位置相同。
图3和图4从电子层面揭示了Si的作用机制。纯TiB/TiAl3界面界面处存在强烈的电荷积累和轨道杂化,形成稳定的Ti-Ti键。而Si掺杂后,电荷在Ti-Si间聚集,形成了强的Ti-Si共价键,这削弱了界面处关键的Ti-Ti相互作用,导致TiAl₃ 2DC稳定性下降。
【总结与展望】
本研究构建了一个多尺度计算框架,CASTEP作为第一性原理计算的引擎,承担了从原子-电子尺度揭示Si中毒微观机理的核心任务。它通过构建并优化TiB₂/TiAl₃/α-Al的系列界面超胞模型,精确计算了清洁与Si掺杂情况下体系的形成能、形核能垒、电荷密度分布以及电子态密度。计算结果表明,Si在TiAl₃二维化合物中的偏聚,会通过形成强Ti-Si共价键,从三个方面导致中毒:一是破坏TiAl₃模板自身的结构有序度与稳定性(PDF与形成能证据),二是削弱其与TiB₂基底的关键界面键合(电荷差分与PDOS证据),三是显著降低其与上方α-Al的化学相互作用并提高形核能垒(PDOS与ΔEα-Al证据)。
基于本研究的成功经验,计算模拟在材料界面工程与合金设计领域拥有广阔前景。首先,可继续深化CASTEP的应用,例如结合AIMD(从头算分子动力学)模拟熔融Al-Si合金与掺杂后TiAl₃2DC界面的动态相互作用过程,直观揭示形核初期的动力学行为;或利用Dmol³模块计算铝液滴在污染界面上的接触角,从润湿性角度量化形核效能的变化。其次,推动高通量计算与机器学习,以CASTEP为主要工具,系统筛查Zr、Nb、V等元素对TiAl₃ 2DC的掺杂效果,计算它们与Si的相互作用能及对界面稳定性的影响,快速筛选出能“抗中毒”的最佳掺杂元素,为设计新一代晶粒细化剂提供理论蓝图。
【文章信息】
原文标题:Insight into Si poisoning on grain refinement of Al–Si/Al–5Ti–B system
标题中文翻译:Si中毒对Al-Si/Al-5Ti-B体系晶粒细化的影响
期刊:《Acta Materialia》
DOI:10.1016/j.actamat.2020.01.039
通讯作者单位:重庆大学 物理科学与工程学院
End

https://wap.sciencenet.cn/blog-3536821-1519674.html
上一篇:Discovery Studio文献解读 | 结核杆菌FtsZ抑制剂计算药理学研究
下一篇:直播预告 | Materials Studio 2026 版本更新亮点