博文
衰老的程序论打败损伤积累论
||
作者:黄必录
摘要:衰老不是由随机损伤并逐渐积累的结果,而是由端粒和rDNA通过P53通路驱动的一种遗传程序。
关键词:衰老的程序论;衰老的损伤积累论;细胞衰老;端粒;45S rDNA;P53
预印本:Huang, Bilu and Hu, Xiaowen, Programmed Aging Theory Defeats Damage Accumulation Theory of Aging (July 28, 2025). Available at SSRN: https://ssrn.com/abstract=5368732
1 介绍
我们每个人都会慢慢变老,然后死亡,而且很多疾病也是由衰老导致的,因此,理解衰老机制至关重要。虽然已提出了超过300种的衰老理论[1],但各种理论都有缺陷,似乎已进入了死胡同,以致于对寿命的干预效果止步不前。根据“DrugAge”每年对延长寿命药物/化合物效果的分析,虽然近年来不断有更多的相关研究,但并未显示出更显著的延长寿命的效果[2]。甚至很多宣称能够延长寿命的抗衰老药都经不起美国国家老龄研究所严格的ITP(Interventions Testing Program) 测试,例如,能增加NAD+的抗衰老药NR,反而使雄性小鼠寿命缩短了3%[3]。因此,抗衰老药如果不能延长寿命,就不能叫抗衰老药,顶多只能叫保健品。
衰老理论分为两种:程序论和损伤积累论。程序论认为,个体的发育和衰老都是由程序控制的。损伤积累论认为,衰老源于分子和细胞的随机损伤并逐渐积累的结果,而修复系统无法完全抵消。该框架包括细胞核DNA和线粒体DNA(mtDNA)的损伤、蛋白质错误折叠或交联变性、以及各种代谢废物的积累。Gyenis等人认为,DNA损伤可能是导致细胞衰老的主要原因[4]。Sinclair等人[5]提出了衰老的信息论(Information Theory of Aging,ITOA),该理论认为,DNA损伤和修复过程中会导致表观遗传信息丢失,从而导致细胞衰老,因此,ITOA也属于衰老的损伤积累论范畴。
2 衰老的损伤积累论不成立
一个正确的理论是不容许存在一个反面的证据,而且反面证据越多越不靠谱,由于衰老的损伤积累论漏洞太多,可以确定该理论是错误的。
2.1 损伤积累不会导致细胞衰老的证据
DNA损伤被认为可能是导致细胞衰老的主要原因[4-5],然而,电离辐射能极大的增加细胞核DNA和mtDNA的损伤,但致死量以下的电离辐射反而能延长果蝇、家蝇、大鼠和小鼠的寿命[6-9]。在日本,原子弹幸存者比平均寿命更长,癌症更少[10]。通常认为,DNA修复能力越强,寿命越长,然而,蟑螂和水熊虫有超强的DNA修复能力,但在条件适宜下,蟑螂和水熊虫寿命只有几个月。
老化的酵母细胞中存在的突变数量相当低。一些具有高水平自由基或突变率的基因工程小鼠品系不会过早衰老,也不会比野生型小鼠寿命更短。小鼠衰老的心肌细胞的细胞核DNA仅有少量的突变[11]。细胞核DNA突变积累并不会加速衰老,但增加了患癌率[12]。HeLa细胞的细胞核会迅速积累DNA损伤[13],然而,HeLa细胞的分裂次数仍然是无限的。
在没有特异性免疫系统又能无性繁殖的植物、涡虫和灯塔水母中,细胞核中的DNA突变的细胞是无法被选择性清除掉的,但并不影响它们无性繁殖和永生性。植物可以用枝条来选育新品种,就是靠分生组织中的干细胞的基因突变,但植物干细胞的表观遗传不会发生衰老[14]。iPSC可检测的DNA损伤超过70%[15],但iPSC的表观遗传也不会衰老[16]。因此,DNA损伤积累不会导致细胞衰老。
氧自由基能使DNA突变、蛋白质交联和变性,然而,线粒体超氧化物歧化酶sod-2基因的缺失,反而延长了秀丽隐杆线虫的寿命,尽管氧化损伤的蛋白质显著增加[17]。线粒体超氧化物歧化酶(SOD2)的杂合突变的小鼠,虽然导致氧化损伤和突变的mtDNA增加,但并没有缩短寿命[18]。小鼠的mtDNA突变和积累不会加速衰老和缩短寿命[19-20]。使用能产生超氧化物和过氧化氢的除草剂“百草枯”处理线虫,线虫的寿命最多可以延长58%。再用抗氧化剂乙酰半胱氨酸处理后,百草枯延长寿命的效果又消失了[21]。亚甲基蓝(MB)是一种很强的抗氧化剂,能渗透到溶酶体和线粒体等细胞器,但经ITP测试发现没能延长小鼠的平均寿命[22]。α- 硫辛酸是一种抗氧化剂,擅长防止脂质过氧化以阻止其有害副产品的积累,这些副产品包括丙烯醛等有害醛类。然而,α- 硫辛酸反而使小鼠中位寿命显著缩短[23]。替普瑞酮(GGA)对哺乳动物组织中的热休克蛋白表达有诱导作用,保证蛋白质能正确折叠,但经ITP测试发现GGA并不能延长小鼠寿命[3]。因此,蛋白质的氧化损伤、错误折叠和DNA突变都不会导致细胞衰老。
受到损伤的线粒体和蛋白质等废物能通过细胞自噬途径降解掉,而白藜芦醇和姜黄素能提高自噬,但经ITP测试发现没能延长小鼠寿命[24]。小鼠卵巢衰老与颗粒细胞自噬增强有关[25]。氯喹(CQ,Chloroquine)是是食品药品监督管理局(FDA)批准的唯一的自噬抑制剂[26],但CQ反而能让大鼠最大寿命延长了13%,这与有史以来在小鼠模型中测试的一些最好的抗衰老药不相上下[27]。关闭老年秀丽隐杆线虫自噬,寿命反而延长了50%[28],增加秀丽隐杆线虫肠道自噬反而会加速衰老[29]。这说明损伤积累并不是想象那样会加速细胞衰老,而且过度增强自噬反而会加速衰老。
2.2 损伤积累是可以克服的
衰老的损伤积累论无法解释寿命只有10几天的秀丽隐杆线虫与400~500百年的北极蛤和格陵兰鲨、人体内寿命只有几天的白细胞与寿命长达几十年的神经元和心肌细胞、一年生植物与寿命长达几千年的植物,它们之间的寿命为何有着如此巨大的差距,这说明损伤可以被修复系统完全抵消掉。
衰老的体细胞可以诱导成年轻的iPSC,生物体在生长发育期可以长期没有损伤积累,人类不再分裂的神经元和心肌细胞的寿命几乎和个体一样长,说明损伤也是可以克服的。例如,突变的mtDNA会通过线粒体自噬和线粒体胞吐(Mitocytosis)途径清除掉[30-31]。在个体水平中,突变的细胞核的DNA(1)可以修复;(2)一旦修复不好就会启动细胞凋亡;(3)如果既修复不好也不发生凋亡,最终也会被免疫监视并清除掉。因此,随着年龄的增长,之所以会逐渐积累DNA突变的细胞,是因为免疫系统的衰老。
脂褐素(LF)是老年斑的主要成分。1973年,Tappel等人将维生素E添加到饲料喂养成年老鼠1年,发现神经元LF确定少见了,但死亡率未见减少[32]。含蛋白质2%的饲料喂养squirrel monkeys(Saimiri scireus)9-15周,神经系统有大量LF形成,然后再喂含蛋白质25%的饲料,又减少了LF[33]。但是,高蛋白饮食反而会加速衰老并缩短寿命[34],因此,LF的积累只是细胞衰老的结果,而非细胞衰老的原因,修复系统能够清除掉已积累的LF。
1958年,Yoshida发现[35],在8小时内,密伊乐藻(Elodoa den.se)有细胞核的原生质体中的叶绿体,经历了衰老和结构破坏的过程,而无细胞核原生质体的叶绿体仍保持绿色和连续积累淀粉。1975年,Wright和Hayflick等人把年轻的细胞核植入去核的衰老的细胞质中,结果细胞恢复了分裂,并按年轻细胞剩余的分裂次数继续分裂下下去,这表明决定细胞衰老的部位是细胞核[36],而不是细胞质。也就是说,只要细胞核是年轻的,细胞质中的LF、突变的mtDNA、错误折叠和交联变性的蛋白质都不会积累到有害的程度。
综上所述,可以确定细胞衰老的损伤积累论是不成立的。
3 衰老是由程序控制的
衰老过程是由程序控制的,还是由随机损伤积累的结果,这是衰老研究中两派激烈争论的问题,前面已经列出很多依据说明衰老的损伤积累论是不成立的。由于每种生物都有一个相对固定的生长发育、成熟衰老和死亡的时间表,这就是一种程序,而且还有更多证据也表明,衰老的本质就是一种程序。
3.1 基因的程序化表达说明衰老是由程序控制的
发育和衰老的过程,基因的表达谱也是逐渐变化的,Gyenis等人和ITOA都认为[4-5],DNA损伤可能是导致基因表达谱变化的原因,然而,随着发育与衰老过程,基因表达谱和DNA甲基化谱的变化都是有序的[37],因此不可能是随机的DNA损伤积累导致的。在小鼠衰老过程中,血浆蛋白中增加的主要是对健康不利的蛋白,减少的主要是对健康有利的蛋白[38],再说损伤是随机和没有方向性的,血浆蛋白组份怎么会有如此规期的变化?这也说明衰老是一种程序。
如果衰老的程序论是对的,在细胞衰老的过程,必然是染色体上的基因群沿着时间轴进行程序化表达的过程,事实也是如此,例如,肝细胞在胚胎期主要表达甲胎蛋白,出生后表达白蛋白,到了老年期,白蛋白基因逐渐关闭,转而表达另一些蛋白[39-41]。由于白蛋白是一种不可缺少的蛋白质,因此,测量白蛋白水平可以预测一个人的死亡期[42-43]。
一个生命从受精卵开始,基因表达谱大致可分为早中晚三种模式:早期基因表达谱主要与胚胎发育有关;中期主要与维持健康和生殖有关;晚期主要与破坏正常生理功能有关。因此,衰老和死亡的过程,就是从有益的基因表达谱逐渐转向有害的基因表达谱的过程[44],晚期基因表达谱是导致阿尔兹海默症、动脉粥样硬化和高血压等等的退行性疾病的发病原因。例如,年轻的巨噬细胞主要表达促进新血管形成的,从而增强组织修复的血管内皮生长因子 A 的异构体VEGF-A165A,而衰老的巨噬细胞主要表达抑制血管形成,从而影响组织修复的血管内皮生长因子 A 的异构体VEGF-A165B[45];清道夫受体(SR-B1)会在衰老的血管内皮细胞中上调,从而主动增加从血液中吸收更多的低密度脂蛋白胆固醇(LDL-C ),促进动脉粥样硬化进展[46];Clusterin (Clu)在衰老的造血干细胞中表达上调,会促使造血干细胞向髓系分化,导致髓系细胞的产生增加[47]。造血干细胞在衰老过程中,大约有1500个基因表达下调,1500个基因表达上调,主要表现为,对健康有害的应激反应和炎症基因随着年龄增长而上调,而对健康有益的染色质重塑和DNA修复基因随着年龄增长而下调[48]。据此,通常的抗衰老思路就是抑制某个上调基因,激活或过表达某个下调基因,但是,这种在代谢层面和信号通路上的干预,只能小幅度延长寿命,而且副作用大,更不可能返老还童,这就是为什么虽然近年来不断有更多的抗衰老药物/化合被发现,但并未显示出更显著延长寿命的原因[2]。
综上提示,寻找导致衰老的根本原因,不是寻找什么基因随着年龄的增长上调或下调,而是要找到这些基因为什么会随着年龄的增长而上调或下调。
3.2 性成熟后衰老仍然是由程序控制的
衰老过程是由程序控制的,还是随机损伤积累的结果,这是衰老研究中两派激烈争论的问题。两派的人都同意,生长发育的速度和性成熟的时间都是由程序控制的。但是在性成熟后,反对衰老是程序控制的人认为,衰老对个体的作用是负面的,由于自然选择只能对个体起作用,因此,个体不可能发展出并且保持对自己不利的程序,衰老只能是身体受到随机发生的损伤逐渐积累的结果。
由于生命经过漫长的演化,已经具备了冗余的克服各种损伤的修复系统,因此,即使在性成熟后,衰老也不可能是由随机发生的损伤逐渐积累的结果。以生活在非洲短命的鳉鱼(或其它物种)为例,可以说明性成熟后衰老仍然是由程序控制的[49]:鳉鱼产的卵会在干季休眠,并在雨季到来形成水塘后再次孵化。在津巴布韦,那里只有短暂的雨季,雨季过后水塘很快干涸,这里的鳉鱼品系(Nothobranchius furzeri)寿命只有3个月,相当于雨季的长度;莫桑比克的雨季比津巴布韦长4倍,那里的鳉鱼品系(Nothobranchius rachovii)可以活9个月;坦桑尼亚的鳉鱼品系(Nothobranchius guentheri)生活在有两个雨季的地方,寿命可以长达16个月。将这三种鳉鱼放在同一条件下进行人工饲养,它们寿命的差别仍然存在[50],这说明衰老是由程序控制的,因为随机损伤积累无法解释这三种同一属的身体结构极为相似的鳉鱼寿命为什么差别如此之大,而且正好与雨季的长度相吻合[51]。
4 控制发育与衰老的程序的运行机制
在一个生命周期里,染色体上大部分基因的序列和拷贝数是固定不变的,因此,要让不变的基因实现程序化表达,就需要一个计时的装置来驱动。由于个体的寿命可以达到百年以上,例如,格陵兰鲨鱼寿命可达400年,因此,候选为驱动遗传程序运行的“计时物质”(相当于沙漏计时器中的沙子)必须非常稳定,没有半衰期。而蛋白质、RNA、mtDNA、以及细胞核DNA和组蛋白的化学修饰,这些都是很不稳定,有半衰期,处于不断降解和补充的动态平衡中,例如,DNA甲基化与去甲基化,组蛋白的乙酰化与去乙酰化都是同时进行的,因此无法形成时间量度,不具备计时物质的属性。也就是说,衰老根本原因不在于RNA、蛋白质、mtDNA和各种表观遗传修饰上。
端粒是由多拷贝的串联重复DNA阵列组成,因此成为计时物质的理想候选者,而且增加端粒长度能显著增加细胞分裂次数,降低衰老标志物[52-53],这是其它的衰老干预措施所达不到的效果,说明端粒缩短是细胞衰老的根本原因之一。但是,有很多物种或同一物种中的有些类型的细胞,端粒不会随着年龄的增长而发生缩短,因此,同样具有多拷贝的串联重复的核糖体DNA(rDNA)阵列,就成为最佳的候选为细胞中第二套的计时物质,于是提出了“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”,本理论认为,端粒和/或rDNA阵列缩短会导致肿瘤抑制蛋白P53水平升高,从而导致细胞衰老(图1)[54]。
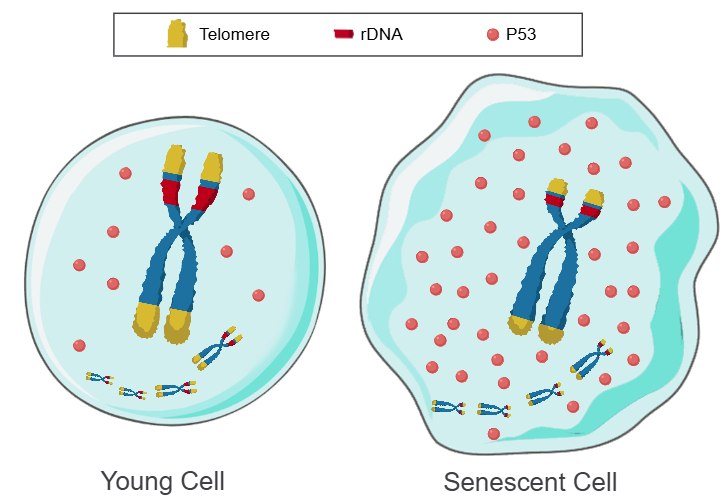
图1:Telomere DNA and ribosomal DNA co-regulation model for cell senescence
a 长阵列的端粒和rDNA,P53迅速降解,细胞年轻。b 短阵列的端粒和rDNA,P53缓慢降解,细胞衰老。
大约1/10的人类基因启动子含有P53结合位点,因此可被归类为P53反应基因[55]。P53不但能够下调总蛋白质合成速率,而且又是一种能同时抑制和激活众多基因的转录因子。因此,随着年龄增长,端粒和/或rDNA阵列逐的渐缩短,P53就会沿着时间轴产生“浓度梯度”,由于P53会与多种基因的启动子和增强子结合,从而使有些基因表达上调,有些基因表达下调,以此驱动染色体上的基因群进行程序化表达[56]。由于不同分化类型的细胞有着不同编程的遗传程序,因此,在衰老过程有着不同的基因表达模式。
5 Telomere DNA and ribosomal DNA co-regulation model for cell senescence的证据
一个理论是否正确,要看这个理论是否具备自洽性,既然端粒DNA和rDNA阵列的缩短是导致细胞衰老的根本原因,是驱动基因群程序化表达的计时物质,那么,在体细胞中损耗掉的端粒DNA和rDNA,在生殖细胞或胚胎的早期细胞中必须要能够补充,否则,生命无法进行世代轮回。幸运的是,已有证据表明,在体细胞中损耗掉的端粒DNA和rDNA,可以在胚胎早期细胞或生殖细胞中得到补充[57-59]。小鼠造血干细胞会因为mTOR1激活导致rDNA阵列缩短[60],而抗衰老药雷帕霉素能通过抑制mTOR1,进而抑制rDNA转录和细胞复制,减缓细胞复制性衰老,延长小鼠寿命。据此,从第一性原理看,物种寿命是由端粒DNA阵列和/或rDNA阵列的缩短速率决定的。
Telomere DNA and ribosomal DNA co-regulation model for cell senescence曾经指出,端粒和/或rDNA阵列缩短会导致肿瘤抑制蛋白P53水平升高,使细胞进入衰老状态,并且多能重编程返老还童机制是因为端粒和rDNA阵列的大幅度延长[54]。据此,我们通过敲低小鼠和人类原代细胞中的 45S rDNA拷贝数,结果:衰老标志物P53、P21、P16和SA-β-GAL都按照预期显著上调,端粒长度、细胞活力和细胞传代次数都显著减少。此外还检测了小鼠的衰老细胞和hESC与hiPSC,发现衰老细胞的端粒长度和45S rDNA拷贝数都显著减少了,hESC与hiPSC的端粒长度和45S rDNA拷贝数显著增加了,这些数据有力的证明了hESC和hiPSC的返老还童机制不是因为表观遗传重编程,而是因为端粒DNA阵列和45S rDNA阵列的长度都显著增加了,细胞衰老和Hayflick极限的根本原因是由端粒和 45S rDNA 共同调控的,而且rDNA对衰老的权重大于端粒(未发表的观察)。
6 端粒和rDNA是非常脆弱的串联重复
Gyenis等人和ITOA都认为[4-5],DNA损伤可能是导致基因表达谱变化的原因,因为在暴露于DNA损伤剂的细胞中,基因表达模式与正常衰老过程中的基因表达模式非常相似。早老症如Cockayne综合征,也以DNA损伤为特征。那么,为什么DNA修复缺陷衰老更快?
Telomere DNA and ribosomal DNA co-regulation model for cell senescence认为[54],DNA损伤的严重程度和/或修复机制的效率可以通过2种途径加速细胞衰老,而不是DNA损伤的本身会导致细胞衰老:(1)端粒DNA和rDNA是属于多拷贝的串联重复DNA,本来稳定性很差,在DNA损伤剂下就更容易导致拷贝数丢失,从而加速了复制性衰老,再说早老症也发现了端粒的快速缩短;(2)DNA损伤的细胞很容易发生凋亡或被免疫系统清除掉,这会刺激周围细胞分裂填补上去,从而加速了细胞的复制性衰老。
7 个体衰老是由成体干细胞的“复制性衰老”导致的
细胞外基质和细胞内的交联变性或错误折叠的蛋白质都是可以降解和更新的,衰老组织之所以会积累这些垃圾,是因为遗传程序关闭了这些降解和更新机制,因此,个体的衰老不可能是大分子垃圾积累导致的[51]。
除了心脏,所有器官组织中都发现了驻留的干细胞,这些干细胞通过自我复制和细胞分化,持续产生新的干细胞和“功能细胞”,维持着组织和器官的稳态与修复。但是,干细胞和功能细都会因为细胞衰老、基因突变或病毒感染等因素而被免疫系统清除掉,为了补充这些损耗掉的细胞,干细胞要经历反复的有丝分裂,由于成体干细胞的分裂次数是有限的,而且每分裂一次就会比上一代更老一些,从而产生了复制性衰老。而由衰老的成本干细胞分化的功能细胞也是衰老的功能细胞,从而导致组织、器官和个体的衰老,因此,导致个体衰老的根本原因,归根结底是由成体干细胞本身的复制性衰老导致的[61]。
如果成体干细胞不会发生复制性衰老。那么,由DNA损伤、细胞毒性化合物和癌基诱导等等因素导致的细胞衰老,并由此损耗掉的成体干细胞和功能细胞,都可以由成体干细胞通过自我复制和细胞分化补充上去,也就是说,如果成体干细胞不会发生复制性衰老,个体就能青春永驻[51]。
8 结语
综上所述,衰老过程是由程序控制的,而不是随机损伤积累的结果。要想逆转个体衰老和大幅度延长寿命,替代器官移植和治愈退行性疾病,唯一的方案就是通过增加组织中的成体干细胞的端粒和rDNA阵列的长度,以此使组织、器官和个体发生返老还童,除此之外,其它的抗衰老措施顶多只能小幅度延长寿命,更不可能返老还童。
参考文献
[1] Medvedev ZA. An attempt at a rational classification of theories of ageing. Biol Rev Camb Philos Soc. 1990 Aug;65(3):375-98. doi: 10.1111/j.1469-185x.1990.tb01428.x.
[2] DrugAge: The Database of Ageing-related Drugs:https://genomics.senescence.info/drugs/index.php
[3] Harrison DE, Strong R, Reifsnyder P, et al. 17-a-estradiol late in life extends lifespan in aging UM-HET3 male mice; nicotinamide riboside and three other drugs do not affect lifespan in either sex. Aging Cell. 2021 May;20(5):e13328. doi: 10.1111/acel.13328. Epub 2021 Mar 31. Erratum in: Aging Cell. 2022 Nov;21(11):e13672. doi: 10.1111/acel.13672.
[4] Gyenis A, Chang J, Demmers JJPG, et al. Genome-wide RNA polymerase stalling shapes the transcriptome during aging. Nat Genet. 2023 Feb;55(2):268-279. doi: 10.1038/s41588-022-01279-6.
[5] Lu YR, Tian X, Sinclair DA. The Information Theory of Aging. Nat Aging. 2023 Dec;3(12):1486-1499. doi: 10.1038/s43587-023-00527-6.
[6] Lamb, MJ. The effects of radiation on the longevity of female Drosophila subobscura. Journal of Insect Physiology, 1964. 10(3): p. 487-497. doi.org/10.1016/0022-1910(64)90072-1.
[7] Allen RG, Sohal RS. Life-lengthening effects of gamma-radiation on the adult housefly, Musca domestica. Mech Ageing Dev. 1982 Dec;20(4):369-75. doi: 10.1016/0047-6374(82)90104-x.
[8] Caratero A, Courtade M, Bonnet L, et al. Effect of a continuous gamma irradiation at a very low dose on the life span of mice. Gerontology. 1998;44(5):272-6. doi: 10.1159/000022024.
[9] Calabrese EJ, Baldwin LA. The effects of gamma rays on longevity. Biogerontology. 2000;1(4):309-19. doi: 10.1023/a:1026510001286.
[10] Sutou S. Low-dose radiation from A-bombs elongated lifespan and reduced cancer mortality relative to un-irradiated individuals. Genes Environ. 2018 Dec 19;40:26. doi: 10.1186/s41021-018-0114-3. Erratum in: Genes Environ. 2019 Apr 19;41:12. doi: 10.1186/s41021-019-0127-6.
[11] De Majo F, Martens L, Hegenbarth JC, et al. Genomic instability in the naturally and prematurely aged myocardium. Proc Natl Acad Sci U S A. 2021 Sep 7;118(36):e2022974118. doi: 10.1073/pnas.2022974118.
[12] Robinson PS, Coorens THH, Palles C, et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat Genet. 2021 Oct;53(10):1434-1442. doi: 10.1038/s41588-021-00930-y.
[13] Liu Y, Mi Y, Mueller T, et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat Biotechnol. 2019 Mar;37(3):314-322. doi: 10.1038/s41587-019-0037-y.
[14] Dai D, Chen K, Tao J, et al. Aging drives a program of DNA methylation decay in plant organs. bioRxiv [Preprint]. 2024 Nov 5:2024.11.04.621941. doi: 10.1101/2024.11.04.621941.
[15] Rouhani FJ, Zou X, Danecek P, et al. Substantial somatic genomic variation and selection for BCOR mutations in human induced pluripotent stem cells. Nat Genet. 2022 Sep;54(9):1406-1416. doi: 10.1038/s41588-022-01147-3.
[16] Koch CM, Reck K, Shao K, et al. Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res. 2013 Feb;23(2):248-59. doi: 10.1101/gr.141945.112.
[17] Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009 Feb;5(2):e1000361. doi: 10.1371/journal.pgen.1000361.
[18] Van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003 Dec 16;16(1):29-37. doi: 10.1152/physiolgenomics.00122.2003.
[19] Vermulst M, Bielas JH, Kujoth GC, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007 Apr;39(4):540-3. doi: 10.1038/ng1988.
[20] Tamashiro H, Ishikawa K, Sadotomo K, et al. Mitochondrial Respiratory Dysfunction Is Not Correlated With Mitochondrial Genotype in Premature Aging Mice. Aging Cell. 2025 May 2:e70085. doi: 10.1111/acel.70085.
[21] Bazopoulou D, Knoefler D, Zheng Y, et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature. 2019 Dec;576(7786):301-305. doi: 10.1038/s41586-019-1814-y.
[22] Harrison DE, Strong R, Allison DB, et al. Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell. 2014 Apr;13(2):273-82. doi: 10.1111/acel.12170.
[23] Farr SA, Price TO, Banks WA, et al. Effect of alpha-lipoic acid on memory, oxidation, and lifespan in SAMP8 mice. J Alzheimers Dis. 2012;32(2):447-55. doi: 10.3233/JAD-2012-120130.
[24] Nadon NL, Strong R, Miller RA, et al. NIA Interventions Testing Program: Investigating Putative Aging Intervention Agents in a Genetically Heterogeneous Mouse Model. EBioMedicine. 2017 Jul;21:3-4. doi: 10.1016/j.ebiom.2016.11.038.
[25] Li F, Zhu J, Liu J, et al. Targeting Estrogen Receptor Beta Ameliorates Diminished Ovarian Reserve via Suppression of the FOXO3a/Autophagy Pathway. Aging Dis. 2024 Feb 25. doi: 10.14336/AD.2024.0221.
[26] Manic G, Obrist F, Kroemer G, et al. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol. 2014 Jul 15;1(1):e29911. doi: 10.4161/mco.29911.
[27] Li W, Zou Z, Cai Y, et al. Low-dose chloroquine treatment extends the lifespan of aged rats. Protein Cell. 2022 Jun;13(6):454-461. doi: 10.1007/s13238-021-00903-1. Erratum in: Protein Cell. 2024 Apr 1;15(4):313. doi: 10.1093/procel/pwad053.
[28] Wilhelm T, Byrne J, Medina R,et al . Neuronal inhibition of the autophagy nucleation complex extends life span in post-reproductive C. elegans. Genes Dev. 2017 Aug 1;31(15):1561-1572. doi: 10.1101/gad.301648.117.
[29] Ezcurra M, Benedetto A, Sornda T, et al. C. elegans Eats Its Own Intestine to Make Yolk Leading to Multiple Senescent Pathologies. Curr Biol. 2018 Aug 20;28(16):2544-2556.e5. doi: 10.1016/j.cub.2018.06.035. Epub 2018 Aug 9. Erratum in: Curr Biol. 2018 Oct 22;28(20):3352. doi: 10.1016/j.cub.2018.10.003.。
[30] Lou G, Palikaras K, Lautrup S, et al. Mitophagy and Neuroprotection. Trends Mol Med. 2020 Jan;26(1):8-20. doi: 10.1016/j.molmed.2019.07.002.
[31] Jiao H, Jiang D, Hu X, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. 2021 May 27;184(11):2896-2910.e13. doi: 10.1016/j.cell.2021.04.027.
[32] Tappel A, Fletcher B, Deamer D. Effect of antioxidants and nutrients on lipid peroxidation fluorescent products and aging parameters in the mouse. J Gerontol. 1973 Oct;28(4):415-24. doi: 10.1093/geronj/28.4.415.
[33] Manocha SL, Sharma SP. Reversibility of lipofuscin accumulation caused by protein malnutrition in the motor cortex of squirrel monkeys, Saimiri scireus. Acta Histochem. 1977;58(2):219-31. doi: 10.1016/S0065-1281(77)80132-3.
[34] van Galen I, Birkisdóttir MB, Ozinga RA, et al. High protein intake causes gene-length-dependent transcriptional decline, shortens lifespan and accelerates ageing in progeroid DNA repair-deficient mice. NPJ Metab Health Dis. 2025 May 22;3:20. doi: 10.1038/s44324-025-00064-3.
[35] Yoshida Y. On some characteristics of the idioblast in Elodea leaf. J Fac Sci, Niigata Univ Ser. II 1958; 2: 173–178.
[36] Wright WE, Hayflick L. Nuclear control of cellular aging demonstrated by hybridization of anucleate and whole cultured normal human fibroblasts. Exp Cell Res. 1975 Nov;96(1):113-21. doi: 10.1016/s0014-4827(75)80043-7.
[37] Bork S, Pfister S, Witt H, et al. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell. 2010 Feb;9(1):54-63. doi: 10.1111/j.1474-9726.2009.00535.x.
[38] Ding J, Kopchick JJ. Plasma biomarkers of mouse aging. Age (Dordr). 2011 Sep;33(3):291-307. doi: 10.1007/s11357-010-9179-z.
[39] [Sigal SH, Brill S, Fiorino AS, Reid LM. The liver as a stem cell and lineage system. Am J Physiol. 1992 Aug;263(2 Pt 1):G139-48. doi: 10.1152/ajpgi.1992.263.2.G139.
[40] Engelhardt NV, Goussev AI, Shipova LJ, et aI. Immunofluorescent study of alpha-foetoprotein (alpha-fp) in liver and liver liver tumours. I. Technique of alpha-fp localization in tissue sections. Int J Cancer. 1971 Mar 15;7(2):198-206. doi: 10.1002/ijc.2910070203.
[41] Camper SA, Tilghman SM. Postnatal repression of the alpha-fetoprotein gene is enhancer independent. Genes Dev. 1989 Apr;3(4):537-46. doi: 10.1101/gad.3.4.537.
[42] Wu CY, Hu HY, Huang N, et al. Albumin levels and cause-specific mortality in community-dwelling older adults. Prev Med. 2018 Jul;112:145-151. doi: 10.1016/j.ypmed.2018.04.015.
[43] Riviati N, Legiran, Indrajaya T, Saleh I, Ali Z, et al. Serum Albumin as Prognostic Marker for Older Adults in Hospital and Community Settings. Gerontol Geriatr Med. 2024 May 7;10:23337214241249914. doi: 10.1177/23337214241249914.
[44] Frenk S, Houseley J. Gene expression hallmarks of cellular ageing. Biogerontology. 2018 Dec;19(6):547-566. doi: 10.1007/s10522-018-9750-z.
[45] Chen M, Chen J, Liu Y, et al. Senescent Macrophages Promote Age-Related Revascularization Impairment by Increasing Antiangiogenic VEGF-A165B Expression. Aging Cell. 2025 Apr 17:e70059. doi: 10.1111/acel.70059.
[46] Huang L, Chambliss KL, Gao X, Yuhanna IS, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019 May;569(7757):565-569. doi: 10.1038/s41586-019-1140-4.
[47] Sun N, Lin CH, Li MY, et al. Clusterin drives myeloid bias in aged hematopoietic stem cells by regulating mitochondrial function. Nat Aging. 2025 Jun 30. doi: 10.1038/s43587-025-00908-z.
[48] Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010 Apr;45(4):286-90. doi: 10.1016/j.exger.2009.12.010.
[49] Dance A. Live fast, die young. Nature. 2016 Jul 21;535(7612):453-5. doi: 10.1038/535453a.
[50] Genade T, Benedetti M, Terzibasi E, et al. Annual fishes of the genus Nothobranchius as a model system for aging research. Aging Cell. 2005 Oct;4(5):223-33. doi: 10.1111/j.1474-9726.2005.00165.x.
[51] Bilu Huang , Xiaowen Hu. Causality of Aging Hallmarks. Aging and disease. 2025 https://doi.org/10.14336/AD.2025.0541
[52] Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998 Jan 16;279(5349):349-52. doi: 10.1126/science.279.5349.349.
[53] Ramunas J, Yakubov E, Brady JJ, et al. Transient delivery of modified mRNA encoding TERT rapidly extends telomeres in human cells. FASEB J. 2015 May;29(5):1930-9. doi: 10.1096/fj.14-259531.
[54] Bilu Huang. Telomere DNA and ribosomal DNA co-regulation model for cell senescence. Negative, 2021. 12(3):9-15. doi:10.13276/j.issn.1674-8913.2021.03.003.
[55] Hoh J, Jin S, Parrado T, et al. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci U S A. 2002 Jun 25;99(13):8467-72. doi: 10.1073/pnas.132268899.
[56] Andrysik Z, Galbraith MD, Guarnieri AL, et al. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017 Oct;27(10):1645-1657. doi: 10.1101/gr.220533.117.
[57] Marion RM, Strati K, Li H, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009 Feb 6;4(2):141-54. doi: 10.1016/j.stem.2008.12.010.
[58] Liu L, Bailey SM, Okuka M, et al. Telomere lengthening early in development. Nat Cell Biol. 2007 Dec;9(12):1436-41. doi: 10.1038/ncb1664.
[59] Lu KL, Nelson JO, Watase GJ, et al. Transgenerational dynamics of rDNA copy number in Drosophila male germline stem cells. Elife. 2018 Feb 13;7:e32421. doi: 10.7554/eLife.32421.
[60] Xu B, Li H, Perry JM, et al. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet. 2017 Jun 22;13(6):e1006771. doi: 10.1371/journal.pgen.1006771.
[61] Bilu Huang. The mechanism significance and treatment of aging[M]. Yanjing Medical Correspondence.1998(7.8):1050-1064.
https://www.researchgate.net/publication/391909829_shuailaodejiliyiyijizhiliao_The_mechanism_significance_and_treatment_of_aging
https://wap.sciencenet.cn/blog-3440171-1496206.html
上一篇:衰老的信息论不靠谱
下一篇:衰老研究的里程碑式进展