博文
神经退行性疾病的根本原因
|
作者:黄必录
摘要:由于神经退行性疾病是由衰老导致的,因此,通过清除不可溶的垃圾蛋白的治疗方案是行不通的。由于细胞衰老是由端粒DNA和rDNA共同调控的,因此,要想治愈神经退行性疾病,最好的办法是通过增加成体干细胞的端粒DNA和rDNA的阵列长度。
关键词:神经退行性疾病;阿尔兹海默症;细胞衰老
强调:
1,清除Aβ没有明显改善AD症状,表明不可溶蛋白质的沉积可能不是导致NDs的发病原因。
2,NDs的发病年龄通常在老年期,表明导致NDs发病的主要原因可能是细胞衰老。
3,大脑中的神经干细胞、骨髓中的造血干细胞和肝脏干细胞的衰老可能是导致AD等退行性疾病的主要原因。
4,端粒和rDNA的阵列缩短可能是导致成体干细胞衰老的根本原因。
本文预印本:Huang, Bilu, The Fundamental Cause of Neurodegenerative Diseases (May 11, 2025). Available at SSRN: https://ssrn.com/abstract=5250307 or http://dx.doi.org/10.2139/ssrn.5250307
介绍
神经退行性疾病(NDs)是一类由进行性神经细胞死亡而导致功能障碍的疾病,包括阿尔茨海默症(AD)、帕金森病(PD)、亨廷顿舞蹈症(HD)、肌萎缩侧索硬化症(ALS)等。NDs的病因尚不明确并且无法治愈,严重威胁着人类健康的同时也造成了巨大的经济负担。
1 在因果关系中,垃圾蛋白质的积累不是导致NDs原因
NDs的大脑中会沉积一些不可溶的垃圾蛋白质,包括β-淀粉样蛋白(Aβ)、超磷酸化tau蛋白(p-Tau)、异常折叠的α-突触核蛋白(α-Syn)和多聚谷氨酰胺蛋白(polyQ)。关于AD的发病原因,1992年,Hardy和Higgins首次提出了淀粉样蛋白级联反应假说,认为脑组织中的Aβ的沉积是AD发病的起始事件,并引发了后续一系列病理变化[1]。近年来又把Aβ和p-Tau与AD和PD联系上[2-5]。
对于一种疾病的治疗,通常的思路是发现什么标志物增加了就进行抑制或清除,什么标志物减少了就进行激活或补充,而不是去思考这些标志物为什么会增加或减少。因此,几十年来主要集中在如何把Aβ清除掉,但是,研究发现,有些患者大脑中有着异常高的Aβ沉积,但并不会导致认识障碍[6],而且清除Aβ未能阻止痴呆的进展[7-10]。最近还发现,Aβ的浓度不管是升高还是降低,都会导致突触生长蛋白(synaptophysin)减少。也就是说,Aβ既不是天使,也不是恶魔,而是一个浓度需要平衡的生理因子,这颠覆了长期以来认为Aβ越少越好的治疗思路[11]。因此,对于AD的治疗,专注于清除p-Tau的结局可能和清除Aβ一样收效甚微。因此,方向不对,努力白费。统计表明,2000年到2017年间,33家顶级制药企业累计在AD领域投入超过6000亿美金全部打水漂了[12]。
2 导致NDs的主要原因是衰老
AD主要见于65岁以上的人[13-15]。ALS通常在40到60岁之间开始发展[16]。HD临床运动发作的平均年龄为40~50岁,其起因是因为某些基因突变导致Huntingtin(HTT)基因第一个外显子内的CAG三个核苷酸序列大量异常重复,该序列编码产生含有多聚谷氨酰胺的蛋白(polyQ),后者可在细胞内异常聚集、形成不可溶的蛋白沉淀。HD有早发(二十岁以前)和晚发(四五十岁左右)两种,其发病年龄的主要决定因素是CAG重复序列的重复次数。然而,这一决定因素仅能够解释60%左右的发病时间差异[17]。因此,HD的发病原因,除了基因突变外,主要原因就是衰老。总之,要想根治NDs,必须要从衰老角度入手。
如上所述,Aβ既不是天使,也不是恶魔,而是一个浓度需要平衡的生理因子,这颠覆了长期以来认为Aβ越少越好的治疗思路[11],而打破这种平衡的根本原因就是衰老,因此,导致NDs的根本原因就是衰老。例如,人类大脑皮层中Aβ前体蛋白(APP)基因启动子区域的甲基化水平会随着年龄的增长而降低,从而提高了该基因的转录活性[18];Aβ主要靠肝脏合成的白蛋白转运到肝脏中降解[19],因此,肝脏衰老会降低对Aβ的清除。肝脏衰老还会上调大脑中的衰老标志物P21[20],而大脑衰老会导致多种参与降解Aβ的酶类表达下调[21-25];小胶质细胞是大脑中的重要的垃圾清洁工,但是,具有负调控小胶质细胞清除垃圾的CD22蛋白的表达会随着衰老而上调,从而降低了小胶质细胞对Aβ等垃圾蛋白的清除[26]。
组织中的功能性终末分化细胞会因各种问题不断损耗掉,然后由组织中的驻留干细胞分化补充。例如,小胶质细胞是一种巨噬细胞,是由骨髓中的造血干细胞分化的[27]。肝脏中的成熟肝细胞是由肝脏干细胞分化的[28]。大脑中的神经元是由神经干细胞分化的。由衰老的成体干细胞分化补充的组织细胞也是衰老的细胞[29],因此,导致AD等NDs的根本原因就是大脑中的神经干细胞和远离大脑的肝脏干细胞和骨髓干细胞衰老导致的[30-32]。
3 细胞衰老是导致NDs的证据
端粒是位于染色体末端的一段多拷贝的串联重复DNA阵列,端粒缩短不但是细胞衰老的一个标志,而且还是导致细胞衰老的主要原因之一,因为增加端粒长度能显著降低衰老标志物和增加细胞的传代次数[33],而干预其它衰老标志物就无法起到这样显著的效果。因此,端粒缩短与AD、PD和HD有关[34-40]。而且在端粒酶缺乏的小鼠,体细胞端粒酶再激活逆转了神经变性,恢复了增殖的Sox2(+)神经祖细胞、Dcx(+)新生神经元和Olig2(+)少突胶质细胞群[41]。
端粒缩短会导致P53上调[42]。P53/P21/P16信号通路在调节衰老方面发挥着重要作用[43]。 约有一半的肿瘤细胞p53基因突变,P53基因缺失可以使细胞无限增殖[44-45],这表明P53是细胞衰老的主控因子。而AD大脑皮质神经元和皮肤有着高水平的P53[46-48],这表明细胞衰老是导致AD的主因。
衰老的细胞会产生一组炎症因子或称为衰老相关分泌表型(Senescence-associated secretory phenotype,SASP)。而P53会激活下游基因p21,P21会通过降低Rb的磷酸化促进SASP的产生[49]。AD患者的神经元也表现出一系列衰老的特征并产生SASP[50]。衰老的细胞在P53的作用下也会产生染色体外的dsRNA和eccDNA,并泄漏到细胞质中[51-52],细胞就会以为感染了病毒而产生免疫反应,产生无菌性炎症和增加Aβ沉积,同时还会表达“来吃我”的信号,吸引免疫细胞进行选择性杀死神经元[53]。
4 驱动NDs进展的原因是基因的程序化表达
如上所述,Aβ既不是天使,也不是恶魔,而是一个浓度需要平衡的生理因子[11],而打破这种平衡的根本原因就是衰老。那么,衰老是如何打破这种平衡的?
由于个体的发育、成熟、衰老和死亡的本质是基因群沿着时间轴进行程序化表达的过程,因此,细胞衰老的过程,必然是染色体上的基因群沿着时间轴进行程序化表达的过程,例如,肝细胞在胚胎期主要表达甲胎蛋白,出生后甲胎蛋白基因逐渐关闭,转而表达白蛋白,到了老年期,白蛋白基因逐渐关闭,转而表达另一些蛋白[54-56]。由于白蛋白是一种缺一不可的蛋白质,因此,测量白蛋白水平可以预测一个人的死亡期[57-58]。
总的来说,衰老不是由随机的损伤和积累驱动的,基因表达也不是一味的下调,而是一种精心设计的遗传程序。过了生殖期,个体的存在已失去了意义,因此,衰老和死亡过程中的基因表达的调控,就是有益基因的逐渐下调和有害基因的逐渐上调的过程[59]。例如,年轻的巨噬细胞主要表达促进新血管形成的血管内皮生长因子 A 的异构体VEGF-A165A,衰老的巨噬细胞主要表达抑制血管形成的VEGF-A165B[60];造血干细胞在衰老过程中,大约有1500个基因表达下调和1500个基因表达上调,主要表现为,应激反应和炎症基因随着年龄增长而上调,而染色质重塑和DNA修复基因随着年龄的增长而下调[61]。
5 基因如何实现程序化表达
如上所述,P53是衰老的主控因子,而且端粒缩短会导致P53上调[42]。转录因子(TF)在不同的基因上具有正向或负向调控作用[62]。大约1/10的人类基因启动子含有P53结合位点[63],因此,P53是一种能够正向和负向调控多种基因转录的TF。因此,随着端粒的逐渐缩短,P53的水平就会延着时间轴逐渐上调,从而驱动一部分基因表达的特异性上调和下调,这样,基因就会呈现出程序化的表达。由于不同分化类型的细胞有着不同编程的遗传程序,因此,在衰老过程有着不同的基因表达模式,例如,造血干细胞在衰老过程中,大约有1500个基因表达下调和1500个基因表达上调[61]。面肩肱型肌营养不良症是一种遗传性的肌肉疾病,DUX4基因的高表达是发病的主要原因,端粒越短,DUX4表达活性越强,随着端粒的逐渐缩短,DUX4表达活性最多上升10倍[64]。DUX4与MHC-I基因表达呈负相关,因此,随着端粒的不断缩短,DUX4表达会逐渐上调,MHC-I表达会逐渐下调,因此,DUX4上调会影响MHC呈递抗原[65],从而增加肿瘤发生率。
综上所述,端粒就是调控遗传病或NDs甚至肿瘤发病的定时炸弹中的计时器。
6 细胞衰老是由端粒DNA和/或rDNA共同调控的
但是,人类某些细胞在衰老过程中端粒不会缩短,细胞复制次数仍然是有限的[66-67],以及用端粒酶维持较长的端粒仍然无法阻止细胞的复制性衰老[68-69]。此外, 酵母菌、 线虫、果蝇和某些鸟类,细胞的端粒也不会缩短[70]。因此,除了端粒DNA,应该还有另一种类似端粒DNA的东西在共同驱动染色体上的基因群沿着时间轴进行程序化表达。为此提出了“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”[71](图1)。
通过敲低小鼠和人类原代细胞中的 45S rDNA拷贝数,结果:衰老标志物P53、P21、P16和SA-β-GAL都显著上调,端粒长度、细胞活力和细胞传代次数都显著减少。此外,还检测了小鼠的衰老细胞和hESC与hiPSC,发现衰老细胞的端粒长度和45S rDNA拷贝数都显著减少了,hESC与hiPSC的端粒长度和45S rDNA拷贝数都显著增加了,这些数据有力的证明了细胞衰老的根本原因是由端粒和 45S rDNA 共同调控的,而且rDNA对衰老的权重大于端粒(Experimental verification on “Telomere DNA and ribosomal DNA co-regulation model for cell senescence”:https://www.biorxiv.org/content/10.1101/2024.07.23.604700v1)。
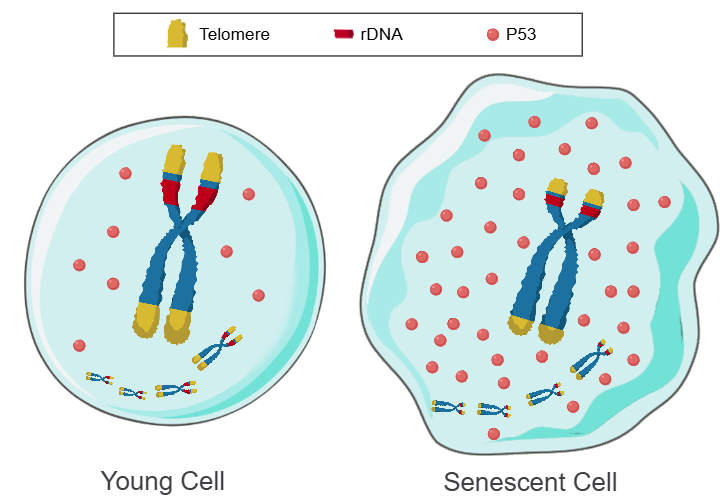
图1:Telomere DNA and ribosomal DNA co-regulation model for cell senescence
a 长阵列的端粒和rDNA,P53迅速降解,细胞年轻。b 短阵列的端粒和rDNA,P53缓慢降解,细胞衰老。
综上所述,个体的生长、发育、成熟、衰老和死亡的本质是一种遗传程序,而程序的运行需要时钟驱动,端粒和rDNA就是驱动遗传程序运行的时钟。也就是说,细胞衰老的根本原因,就是端粒DNA阵列和/或rDNA阵列的缩短,并通过P53介导染色体上的基因群沿着时间轴进行程序化表达[72],例如,已发现不同长度的端粒DNA阵列和rDNA阵列或不同浓度的P53能通过影响表观遗传来调控众多个基因表达[73-79]。据此,要想治愈AD等NDs和大幅度延长寿命,唯一的方案就是通过增加成体干细胞中的端粒DNA阵列和rDNA阵列的长度,除此之外,其它的抗衰老措施都是徒劳的。
一个理论是否正确,要看这个理论是否具备自洽性,既然端粒DNA和rDNA是调控细胞衰老的根本原因,是驱动基因群程序化表达的计时物质,那么,在体细胞中损耗掉的端粒DNA和rDNA,在生殖细胞或胚胎的早期细胞中必须要能够补充[80],否则,生命无法进行世代轮回。幸运的是,已有证据表明,在体细胞中损耗掉的端粒DNA和rDNA,可以在胚胎早期细胞或生殖细胞中得到补充[81-83],检测也发现,hESC和hiPSC的返老还童机制不是因为表观遗传重编程,而是因为端粒DNA阵列和45S rDNA阵列的长度都显著增加了(Experimental verification on “Telomere DNA and ribosomal DNA co-regulation model for cell senescence”:https://www.biorxiv.org/content/10.1101/2024.07.23.604700v1),因此,“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”是唯一高度可信的细胞衰老理论。
7 主要衰老理论的缺陷
由于解释生物为什么会衰老的理论太多太乱,因此,在这里,我想对一此主要的衰老理论进行评述。
很多看似很有道理的主流衰老理论都不尽人意,例如,细胞核DNA突变和积累,被很多人认为是导致细胞衰老的主要原因。然而,自然老化的小鼠,心肌细胞的细胞核很少积累突变的DNA[84]。这项研究发现,人类细胞核积累突变的DNA不会加速衰老[85]。HeLa细胞的细胞核会迅速积累DNA损伤[86],然而,HeLa细胞的复制次数仍然是无限的。综上所述,细胞衰老不是由细胞核的DNA突变和积累导致的。
线粒体DNA(mtDNA)的突变和积累也被认为是导致细胞衰老的主要原因[87]。然而,Vermulst等人[88] 发现,增加mtDNA突变的小鼠模型没有任何加速衰老的迹象,也不会缩短寿命。超氧化物歧化酶(SOD2)的杂合突变虽然导致氧化损伤和mtDNA突变增加,但并没有缩短小鼠的寿命[89]。综上所述,细胞衰老不是由mtDNA突变和积累导致的。
衰老的表观遗传理论[90],也是漏洞百出,因为将老年成纤维细胞直接重编程或称转分化为神经干细胞,仍然保留了衰老的表型[91],提示,通过转分化途径治疗NDs是不可行的。在重编程过程中,细胞生理状态的年轻化发生在表观遗传特征的年轻化之前[92],说明重编程导致的衰老逆转可能不是来自于表观遗传重编程。部分重编程结束后,发现端粒没有延长或略有缩短,衰老标志物又开始积累,表观遗传年龄又恢复到重编程之前的状态[93]。部分重编程仅增加野生型小鼠中位寿命12% [94],还不如小分子抗衰老药。由于秀丽隐杆线虫等少数动物中并不存在DNA胞嘧啶甲基化现象[95],因此,加州大学洛杉矶分校,在表观遗传时钟领域有诸多研究的Steve Horvath教授说:“美国国立卫生研究院一直支持我,但还不够,因为一些研究人员认为,由于秀丽隐杆线虫没有表观遗传时钟,它怎么会衰老?”。
结语
总之,导致AD等NDs的根本原因,主要是神经干细胞、造血干细胞和肝脏干细胞的端粒和rDNA阵列缩短导致的。
参考文献
[1] Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992 Apr 10;256(5054):184-5. doi: 10.1126/science.1566067.
[2] Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer's disease. Nat Neurosci. 2020 Oct;23(10):1183-1193. doi: 10.1038/s41593-020-0687-6.
[3] Pang K, Jiang R, Zhang W, et al. An App knock-in rat model for Alzheimer's disease exhibiting Aβ and tau pathologies, neuronal death and cognitive impairments. Cell Res. 2022 Feb;32(2):157-175. doi: 10.1038/s41422-021-00582-x.
[4] Düzel E, Ziegler G, Berron D, et al. Amyloid pathology but not APOE ε4 status is permissive for tau-related hippocampal dysfunction. Brain. 2022 May 24;145(4):1473-1485. doi: 10.1093/brain/awab405.
[5] Chu Y, Hirst WD, Federoff HJ, et al. Nigrostriatal tau pathology in parkinsonism and Parkinson's disease. Brain. 2024 Feb 1;147(2):444-457. doi: 10.1093/brain/awad388. Erratum in: Brain. 2025 Apr 29:awaf146. doi: 10.1093/brain/awaf146.
[6] Arboleda-Velasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. doi: 10.1038/s41591-019-0611-3.
[7] Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008 Jul 19;372(9634):216-23. doi: 10.1016/S0140-6736(08)61075-2.
[8] van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in Early Alzheimer's Disease. N Engl J Med. 2023 Jan 5;388(1):9-21. doi: 10.1056/NEJMoa2212948.
[9]Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer's Disease. J Prev Alzheimers Dis. 2022;9(2):197-210. doi: 10.14283/jpad.2022.30.
[10] Sims JR, Zimmer JA, Evans CD, et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 2023 Aug 8;330(6):512-527. doi: 10.1001/jama.2023.13239.
[11] McGeachan RI, Meftah S, Taylor LW, et al. Divergent actions of physiological and pathological amyloid-β on synapses in live human brain slice cultures. Nat Commun. 2025 Apr 30;16(1):3753. doi: 10.1038/s41467-025-58879-z.
[12] New Report Expresses Optimism Over Current State of Alzheimer's Drug Development. Retrieved December 4, 2018, from
https://www.biospace.com/article/pharma-industry-report-on-alzheimer-s-research-in-the-u-s-cautiously-optimistic/
[13] Brookmeyer R, Johnson E, Ziegler-Graham K, et al. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. 2007 Jul;3(3):186-91. doi: 10.1016/j.jalz.2007.04.381.
[14] Lebert F, Leroy M, Pasquier F, Strubel D. Young onset demented patients in French cognitive-behavioral specialized units. Geriatr Psychol Neuropsychiatr Vieil. 2016 Jun 1;14(2):194-200. English. doi: 10.1684/pnv.2016.0607.
[15] Wilson RS, Wang T, Yu L, et al. Cognitive Activity and Onset Age of Incident Alzheimer Disease Dementia. Neurology. 2021 Aug 31;97(9):e922-e929. doi: 10.1212/WNL.0000000000012388.
[16] Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006 Oct 5;52(1):39-59. doi: 10.1016/j.neuron.2006.09.018. PMID: 17015226.
[17] Handsaker RE, Kashin S, Reed NM, et al. Long somatic DNA-repeat expansion drives neurodegeneration in Huntington's disease. Cell. 2025 Feb 6;188(3):623-639.e19. doi: 10.1016/j.cell.2024.11.038.
[18] Tohgi H, Utsugisawa K, Nagane Y, et al. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999 Jul 5;70(2):288-92. doi: 10.1016/s0169-328x(99)00163-1.
[19] Cheng Y, He CY, Tian DY, et al. Physiological β-amyloid clearance by the liver and its therapeutic potential for Alzheimer's disease. Acta Neuropathol. 2023 Jun;145(6):717-731. doi: 10.1007/s00401-023-02559-z.
[20] Kiourtis C, Terradas-Terradas M, Gee LM, et al. Hepatocellular senescence induces multi-organ senescence and dysfunction via TGFβ. Nat Cell Biol. 2024 Dec;26(12):2075-2083. doi: 10.1038/s41556-024-01543-3. Epub 2024 Nov 13. PMID: 39537753; PMCID: PMC11628396.
[21] Apelt J, Ach K, Schliebs R. Aging-related down-regulation of neprilysin, a putative beta-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of beta-amyloid plaques. Neurosci Lett. 2003 Mar 27;339(3):183-6. doi: 10.1016/s0304-3940(03)00030-2.
[22] Yasojima K, Akiyama H, McGeer EG, et al. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of beta-amyloid peptide. Neurosci Lett. 2001 Jan 12;297(2):97-100. doi: 10.1016/s0304-3940(00)01675-x.
[23] Cook DG, Leverenz JB, McMillan PJ, et al. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer's disease is associated with the apolipoprotein E-epsilon4 allele. Am J Pathol. 2003 Jan;162(1):313-9. doi: 10.1016/s0002-9440(10)63822-9.
[24] Pacheco-Quinto J, Herdt A, et al. Endothelin-converting enzymes and related metalloproteases in Alzheimer's disease. J Alzheimers Dis. 2013;33 Suppl 1(0 1):S101-10. doi: 10.3233/JAD-2012-129043.
[25] Hellström-Lindahl E, Ravid R, Nordberg A. Age-dependent decline of neprilysin in Alzheimer's disease and normal brain: inverse correlation with A beta levels. Neurobiol Aging. 2008 Feb;29(2):210-21. doi: 10.1016/j.neurobiolaging.2006.10.010.
[26] Pluvinage JV, Haney MS, Smith BAH, et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature. 2019 Apr;568(7751):187-192. doi: 10.1038/s41586-019-1088-4.
[27] Yoo Y, Neumayer G, Shibuya Y, et al. A cell therapy approach to restore microglial Trem2 function in a mouse model of Alzheimer's disease. Cell Stem Cell. 2023 Aug 3;30(8):1043-1053.e6. doi: 10.1016/j.stem.2023.07.006. Erratum in: Cell Stem Cell. 2023 Oct 5;30(10):1392. doi: 10.1016/j.stem.2023.08.011.
[28] Lin S, Nascimento EM, Gajera CR, et al. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature. 2018 Apr;556(7700):244-248. doi: 10.1038/s41586-018-0004-7.
[29] Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010 Mar 25;464(7288):520-8. doi: 10.1038/nature08982.
[30] Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer's disease. Nat Med. 2019 Apr;25(4):554-560. doi: 10.1038/s41591-019-0375-9.
[31] Palmer TD, Schwartz PH, Taupin P, et al. Cell culture. Progenitor cells from human brain after death. Nature. 2001 May 3;411(6833):42-3. doi: 10.1038/35075141.
[32] 黄必录.探讨骨髓干细胞衰老与阿尔茨海默病等衰老性疾病的关系[J].医学理论与实践, 2022 ,35 (6 ):933-935. doi: 10.19381/j.issn.1001-7585.2022.06.010.
[33] Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998 Jan 16;279(5349):349-52. doi: 10.1126/science.279.5349.349.
[34] Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer's disease. Neuromolecular Med. 2013 Mar;15(1):25-48. doi: 10.1007/s12017-012-8207-9.
[35] Flanary BE, Sammons NW, Nguyen C, et al. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007 Mar;10(1):61-74. doi: 10.1089/rej.2006.9096.
[36] Kota LN, Bharath S, Purushottam M, et al. Reduced telomere length in neurodegenerative disorders may suggest shared biology. J Neuropsychiatry Clin Neurosci. 2015;27(2):e92-6. doi: 10.1176/appi.neuropsych.13100240.
[37] Maeda T, Guan JZ, Koyanagi M, et al. Aging-associated alteration of telomere length and subtelomeric status in female patients with Parkinson's disease. J Neurogenet. 2012 Jun;26(2):245-51. doi: 10.3109/01677063.2011.651665.
[38] Scarabino D, Veneziano L, Peconi M, et al. Leukocyte telomere shortening in Huntington's disease. J Neurol Sci. 2019 Jan 15;396:25-29. doi: 10.1016/j.jns.2018.10.024.
[39] PerezGrovas-Saltijeral A, Ochoa-Morales A, Miranda-Duarte A, et al. Telomere length analysis on leukocytes derived from patients with Huntington Disease. Mech Ageing Dev. 2020 Jan;185:111189. doi: 10.1016/j.mad.2019.111189.
[40] Niu B, Wu JX, Huang XL, et al. Telomere Length Is a Driving Hallmark for Aging-Related Biochemical Hallmarks: Evidence From the Shared Genetic Effect and Causal Inference. J Gerontol A Biol Sci Med Sci. 2024 Apr 1;79(4):glad275. doi: 10.1093/gerona/glad275.
[41] Jaskelioff M, Muller FL, Paik JH, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011 Jan 6;469(7328):102-6. doi: 10.1038/nature09603.
[42] Leri A, Franco S, Zacheo A, et al. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 2003 Jan 2;22(1):131-9. doi: 10.1093/emboj/cdg013.
[43] Yang F, Yi M, Liu Y, et al. Glutaredoxin-1 Silencing Induces Cell Senescence via p53/p21/p16 Signaling Axis. J Proteome Res. 2018 Mar 2;17(3):1091-1100. doi: 10.1021/acs.jproteome.7b00761.
[44] Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991 Sep;196(1):33-9. doi: 10.1016/0014-4827(91)90453-2.
[45] Navalkar A, Paul A, Sakunthala A, et al. Oncogenic gain of function due to p53 amyloids occurs through aberrant alteration of cell cycle and proliferation. J Cell Sci. 2022 Aug 1;135(15):jcs259500. doi: 10.1242/jcs.259500.
[46] Cenini G, Sultana R, Memo M, et al. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic Biol Med. 2008 Jul 1;45(1):81-5. doi: 10.1016/j.freeradbiomed.2008.03.015.
[47] Cenini G, Sultana R, Memo M, et al. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer's disease. J Cell Mol Med. 2008 Jun;12(3):987-94. doi: 10.1111/j.1582-4934.2008.00163.x.
[48] Lanni C, Uberti D, Racchi M, et al. Unfolded p53: a potential biomarker for Alzheimer's disease. J Alzheimers Dis. 2007 Aug;12(1):93-9. doi: 10.3233/jad-2007-12109.
[49] Sturmlechner I, Zhang C, Sine CC, et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science. 2021 Oct 29;374(6567):eabb3420. doi: 10.1126/science.abb3420.
[50] Herdy JR, Traxler L, Agarwal RK, et al. Increased post-mitotic senescence in aged human neurons is a pathological feature of Alzheimer's disease. Cell Stem Cell. 2022 Dec 1;29(12):1637-1652.e6. doi: 10.1016/j.stem.2022.11.010.
[51] Victorelli S, Salmonowicz H, Chapman J, et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature. 2023 Oct;622(7983):627-636. doi: 10.1038/s41586-023-06621-4. Epub 2023 Oct 11. Erratum in: Nature. 2024 Jan;625(7995):E15. doi: 10.1038/s41586-023-07002-7.
[52] Zhou X, Singh M, Sanz Santos G, et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021 Dec 1;11(12):3090-3105. doi: 10.1158/2159-8290.CD-20-1741.
[53] Zalocusky KA, Najm R, Taubes AL, et al. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer's disease. Nat Neurosci. 2021 Jun;24(6):786-798. doi: 10.1038/s41593-021-00851-3.
[54] Sigal SH, Brill S, Fiorino AS, Reid LM. The liver as a stem cell and lineage system. Am J Physiol. 1992 Aug;263(2 Pt 1):G139-48. doi: 10.1152/ajpgi.1992.263.2.G139. PMID: 1325126.
[55] Engelhardt NV, Goussev AI, Shipova LJ, et aI. Immunofluorescent study of alpha-foetoprotein (alpha-fp) in liver and liver liver tumours. I. Technique of alpha-fp localization in tissue sections. Int J Cancer. 1971 Mar 15;7(2):198-206. doi: 10.1002/ijc.2910070203.
[56] Camper SA, Tilghman SM. Postnatal repression of the alpha-fetoprotein gene is enhancer independent. Genes Dev. 1989 Apr;3(4):537-46. doi: 10.1101/gad.3.4.537.
[57] Wu CY, Hu HY, Huang N, et al. Albumin levels and cause-specific mortality in community-dwelling older adults. Prev Med. 2018 Jul;112:145-151. doi: 10.1016/j.ypmed.2018.04.015.
[58] Riviati N, Legiran, Indrajaya T, Saleh I, Ali Z, et al. Serum Albumin as Prognostic Marker for Older Adults in Hospital and Community Settings. Gerontol Geriatr Med. 2024 May 7;10:23337214241249914. doi: 10.1177/23337214241249914.
[59] Frenk S, Houseley J. Gene expression hallmarks of cellular ageing. Biogerontology. 2018 Dec;19(6):547-566. doi: 10.1007/s10522-018-9750-z.
[60] Chen M, Chen J, Liu Y, et al. Senescent Macrophages Promote Age-Related Revascularization Impairment by Increasing Antiangiogenic VEGF-A165B Expression. Aging Cell. 2025 Apr 17:e70059. doi: 10.1111/acel.70059.
[61] Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010 Apr;45(4):286-90. doi: 10.1016/j.exger.2009.12.010.
[62] Kang Y, Patel NR, Shively C, et al. Dual threshold optimization and network inference reveal convergent evidence from TF binding locations and TF perturbation responses. Genome Res. 2020 Mar;30(3):459-471. doi: 10.1101/gr.259655.119.
[63] Hoh J, Jin S, Parrado T, et al. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci U S A. 2002 Jun 25;99(13):8467-72. doi: 10.1073/pnas.132268899.
[64] Stadler G, Rahimov F, King OD,et al. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat Struct Mol Biol. 2013 Jun;20(6):671-8. doi: 10.1038/nsmb.2571.
[65] Chew GL, Campbell AE, De Neef E, et al. DUX4 Suppresses MHC Class I to Promote Cancer Immune Evasion and Resistance to Checkpoint Blockade. Dev Cell. 2019 Sep 9;50(5):658-671.e7. doi: 10.1016/j.devcel.2019.06.011.
[66] Egan CA, Savre-Train I, Shay JW, et al. Analysis of telomere lengths in human corneal endothelial cells from donors of different ages. Invest Ophthalmol Vis Sci. 1998 Mar;39(3):648-53.
[67] Demanelis K, Jasmine F, Chen LS, et al. Determinants of telomere length across human tissues. Science. 2020 Sep 11;369(6509):eaaz6876. doi: 10.1126/science.aaz6876.
[68] MacKenzie KL, Franco S, May C, et al. Mass cultured human fibroblasts overexpressing hTERT encounter a growth crisis following an extended period of proliferation. Exp Cell Res. 2000 Sep 15;259(2):336-50. doi: 10.1006/excr.2000.4982.
[69] Martin JA, Mitchell CJ, Klingelhutz AJ, et al. Effects of telomerase and viral oncogene expression on the in vitro growth of human chondrocytes. J Gerontol A Biol Sci Med Sci. 2002 Feb;57(2):B48-53. doi: 10.1093/gerona/57.2.b48.
[70] Haussmann MF, Winkler DW, O'Reilly KM, et al. Telomeres shorten more slowly in long-lived birds and mammals than in short-lived ones. Proc Biol Sci. 2003 Jul 7;270(1522):1387-92. doi: 10.1098/rspb.2003.2385.
[71] Bilu Huang.Telomere DNA and ribosomal DNA co-regulation model for cell senescence[J].Negative,2021,12(3):9-15.doi:10.13276/j.issn.1674-8913.2021.03.003(2021).
[72] Andrysik Z, Galbraith MD, Guarnieri AL, et al. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017 Oct;27(10):1645-1657. doi: 10.1101/gr.220533.117.
[73] Demanelis K, Jasmine F, Chen LS, et al. Determinants of telomere length across human tissues. Science. 2020 Sep 11;369(6509):eaaz6876. doi: 10.1126/science.aaz6876.
[74] Carlund O, Norberg A, Osterman P, et al. DNA methylation variations and epigenetic aging in telomere biology disorders. Sci Rep. 2023 May 16;13(1):7955. doi: 10.1038/s41598-023-34922-1.
[75] Larson K, Yan SJ, Tsurumi A, et al. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet. 2012 Jan;8(1):e1002473. doi: 10.1371/journal.pgen.1002473.
[76] Paredes S, Maggert KA. Ribosomal DNA contributes to global chromatin regulation. Proc Natl Acad Sci U S A. 2009 Oct 20;106(42):17829-34. doi: 10.1073/pnas.0906811106.
[77] Andrysik Z, Galbraith MD, Guarnieri AL, et al. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017 Oct;27(10):1645-1657. doi: 10.1101/gr.220533.117.
[78] Peterson EJ, Bögler O, Taylor SM. p53-mediated repression of DNA methyltransferase 1 expression by specific DNA binding. Cancer Res. 2003 Oct 15;63(20):6579-82.
[79] Challen GA, Sun D, Jeong M,et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011 Dec 4;44(1):23-31. doi: 10.1038/ng.1009.
[80] 黄必录.个体发育和端粒的关系.中国老年病杂志[J],2008,5(1):57-58. https://lab.rejubio.cn/static/PDF/The-relationship-between-ontogenesis-and-telomeres.pdf
[81] Marion RM, Strati K, Li H, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009 Feb 6;4(2):141-54. doi: 10.1016/j.stem.2008.12.010.
[82] Liu L, Bailey SM, Okuka M, et al. Telomere lengthening early in development. Nat Cell Biol. 2007 Dec;9(12):1436-41. doi: 10.1038/ncb1664.
[83] Lu KL, Nelson JO, Watase GJ, et al. Transgenerational dynamics of rDNA copy number in Drosophila male germline stem cells. Elife. 2018 Feb 13;7:e32421. doi: 10.7554/eLife.32421.
[84] De Majo F, Martens L, Hegenbarth JC, et al. Genomic instability in the naturally and prematurely aged myocardium. Proc Natl Acad Sci U S A. 2021 Sep 7;118(36):e2022974118. doi: 10.1073/pnas.2022974118.
[85] Robinson PS, Coorens THH, Palles C, et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat Genet. 2021 Oct;53(10):1434-1442. doi: 10.1038/s41588-021-00930-y.
[86] Liu Y, Mi Y, Mueller T, et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat Biotechnol. 2019 Mar;37(3):314-322. doi: 10.1038/s41587-019-0037-y.
[87] Miquel J, Economos AC, Fleming J, et al. Mitochondrial role in cell aging. Exp Gerontol. 1980;15(6):575-91. doi: 10.1016/0531-5565(80)90010-8.
[88] Vermulst M, Bielas JH, Kujoth GC, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007 Apr;39(4):540-3. doi: 10.1038/ng1988.
[89] Van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003 Dec 16;16(1):29-37. doi: 10.1152/physiolgenomics.00122.2003.
[90] Lu YR, Tian X, Sinclair DA. The Information Theory of Aging. Nat Aging. 2023 Dec;3(12):1486-1499. doi: 10.1038/s43587-023-00527-6.
[91] Mertens J, Paquola ACM, Ku M, et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell. 2015 Dec 3;17(6):705-718. doi: 10.1016/j.stem.2015.09.001.
[92] Zhang W, Qu J, Liu GH, et al. The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Biol. 2020 Mar;21(3):137-150. doi: 10.1038/s41580-019-0204-5.
[93] Gill D, Parry A, Santos F, et al. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. Elife. 2022 Apr 8;11:e71624. doi: 10.7554/eLife.71624.
[94] Sahu SK, Reddy P, Lu J, et al. Targeted partial reprogramming of age-associated cell states improves markers of health in mouse models of aging. Sci Transl Med. 2024 Sep 11;16(764):eadg1777. doi: 10.1126/scitranslmed.adg1777.
[95] Suzuki MM, Kerr AR, De Sousa D, et a. CpG methylation is targeted to transcription units in an invertebrate genome. Genome Res. 2007 May;17(5):625-31. doi: 10.1101/gr.6163007.
https://wap.sciencenet.cn/blog-3440171-1494262.html
上一篇:肿瘤机制与治疗策略
下一篇:衰老的信息论不靠谱