
 精选
精选
生理学家的生理学家
奥古斯特·克罗格关于肌肉微血管控制和氧气输送的理论:基于新数据的范式转变
摘要
奥古斯特·克罗格曾两次获得享有盛誉的国际斯特egen奖,分别是因为氮代谢研究(1906年)和推翻气体通过肺上皮细胞主动运输的概念(1910年)。尽管如此,在1920年初,这位技艺精湛的实验家在世界范围内甚至在他自己所在的哥本哈根大学教职员工中都相对默默无闻。但是,在1919年初,他向当时英国《生理学杂志》的编辑兰利博士提交了三篇论文。这些论文将骨骼肌毛细血管数量的解剖学观察与氧气扩散理论相结合,提出了毛细血管的一种新的主动作用,解释了从休息到运动时血液 - 肌肉氧气通量的巨大增加。尽管克罗格自己对他的朋友——杰出的剑桥呼吸生理学家约瑟夫·巴克罗格夫特评价前两篇论文 “相当枯燥”,但他认为第三篇关于氧气供应和毛细血管调节的论文 “很有趣”。这些论文为克罗格在1920年毫无争议地赢得了诺贝尔生理学或医学奖,也构成了这篇综述的基础。它们独自将毛细血管的作用从被动的导管和交换血管(其功能受上游小动脉的支配)转变为独立的收缩单位,这些单位在休息时主要是关闭的,而在肌肉收缩时则主动开放,他将这一过程称为 “毛细血管募集”。在本文中,我们研究了克罗格的发现以及他面临的一些实验困难。特别是,他为模型选择的边界条件(例如,深度麻醉的动物、可忽略不计的肌细胞内氧气分压、毛细血管的二元开闭功能)未能经受住时间的考验。随后,我们用中间的发现来更新读者的认识,这些发现支撑了我们目前对肌肉微循环控制的理解,并回顾了克罗格的发现。本文提出了一种观点,即诺贝尔奖的认可在这种情况下可能导致科学家们忽视了令人信服的证据。就像他和玛丽·克罗格证明了气体通过血气屏障的主动运输在肺部是不必要的一样,骨骼肌中的毛细血管也不会自发或主动地开放和关闭,而且这对于解释运动时血液 - 肌肉氧气通量的增加也不是必要的。因此,当代的毛细血管功能模型的特点是大多数肌肉毛细血管在休息时就支持血液流动,并且从休息到运动时,血液 - 肌细胞氧气通量的增加主要不是通过毛细血管主动扩张来实现的,而是通过提高已经有血液流动的毛细血管中的红细胞和血浆通量来实现的。克罗格因其作为实验家的卓越才华以及提出了导致富有成效的研究途径(包括微血管功能研究)的科学问题而受到赞誉。
引言:“生理学家的生理学家”
沙克·奥古斯特·斯滕伯格·克罗格(1874 - 1949)以其在多个科学领域的开创性发现而闻名,包括生理学、比较生理学、运动生理学、新陈代谢、病理学和昆虫学(《美国医学会杂志》社论,1967年)。作为一名专注的教育家和有才华的教师(克罗格,1929年,1939年)以及技艺精湛的实验家,克罗格被许多人视为比较生理学之父(克罗格,1941年;坦尼,1996年)。然而,谦逊的克罗格本人可能会反对在他死后将他的名字(即克罗格原理)与克劳德·伯纳德1865年的精彩观察联系起来:“对于如此众多的问题,会有某种选择的动物,或者少数这样的动物,在它们身上可以最方便地进行研究”(伯纳德,1865年)。
此外,克罗格与阿奇博尔德·希尔和奥托·迈耶霍夫(1922年诺贝尔生理学奖获得者)等研究人员一起,被公认为现代运动生理学的先驱。而且,他在诸如水生动物的食物供应、动物的渗透调节、离子和水的跨膜运输、生物学研究中的同位素以及昆虫飞行生理学等不同领域发起了广泛的研究和研究方法。在当前气候变化的时代,克罗格1904年关于格陵兰探险中海水二氧化碳溶解度的报告(克罗格,1904年)对于大气二氧化碳命运的预测仍然有效。出于对自然和发现的无尽好奇心,他的才华将卓越的工程技能与对观察和测量准确性的偏好结合在一起。在他的大部分科学生涯中,他最信任的科学知己和同事是他的妻子玛丽(娘家姓乔根森);她是一位有先见之明的科学家和积极主动的医生,她的原创技术和见解至今仍为肺生理学和医学提供着信息。克罗格的一项卓越能力是将设计、构建和测试他所需的测量仪器作为一种思维练习(施密特 - 尼尔森,1984年,1995年)。例如,他的微压计用于测量呼出气体或血液中的气体分压,其精度高达惊人的0.001%(克罗格,1920年a):这一能力超过了当今许多市售的分析仪!此外,克罗格在自己的实验室车间制造的记录式肺活量计、自行车测力计、精密移液器和呼吸装置在世界各地的医院和实验室中得到了广泛使用。
本文的主要目的是回顾克罗格在《生理学杂志》上发表的三篇论文(克罗格,1919年a、b、c),这些论文涉及肌肉毛细血管功能和肌肉的氧气供应,为他赢得了1920年的诺贝尔生理学或医学奖。毫无疑问,这些论文与当时关于微循环的解剖学和生理学以及其在血液 - 肌肉氧气通量中允许作用的流行观点大相径庭。克罗格巧妙地将他的氧气扩散测量和理论与对肌肉毛细血管红细胞(RBC)流动的观察(主要是在青蛙中)以及对豚鼠和其他物种的毛细血管计数相结合,提出了他的 “毛细血管运动” 机制,用于控制肌肉灌注氧气输送()以及休息和运动时肌细胞内的氧气分压(图1,左;另见克罗格,1920年c)。以前,毛细血管在很大程度上被认为是被动的导管和交换血管,血管阻力以及毛细血管红细胞和血浆通量的控制取决于上游的动脉/小动脉。
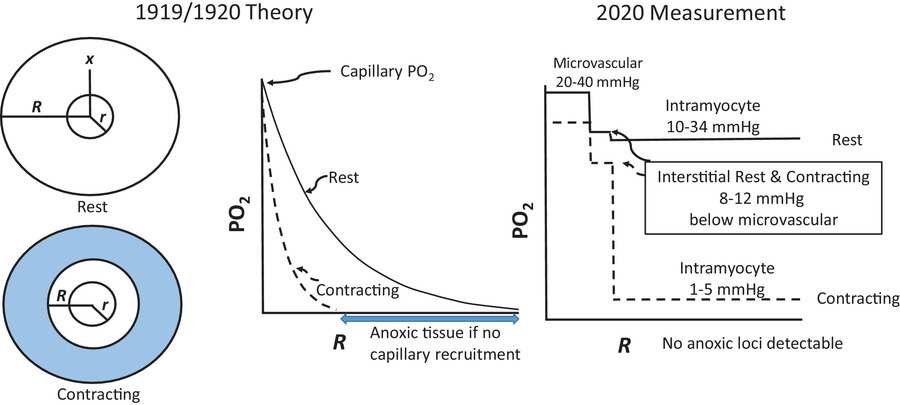
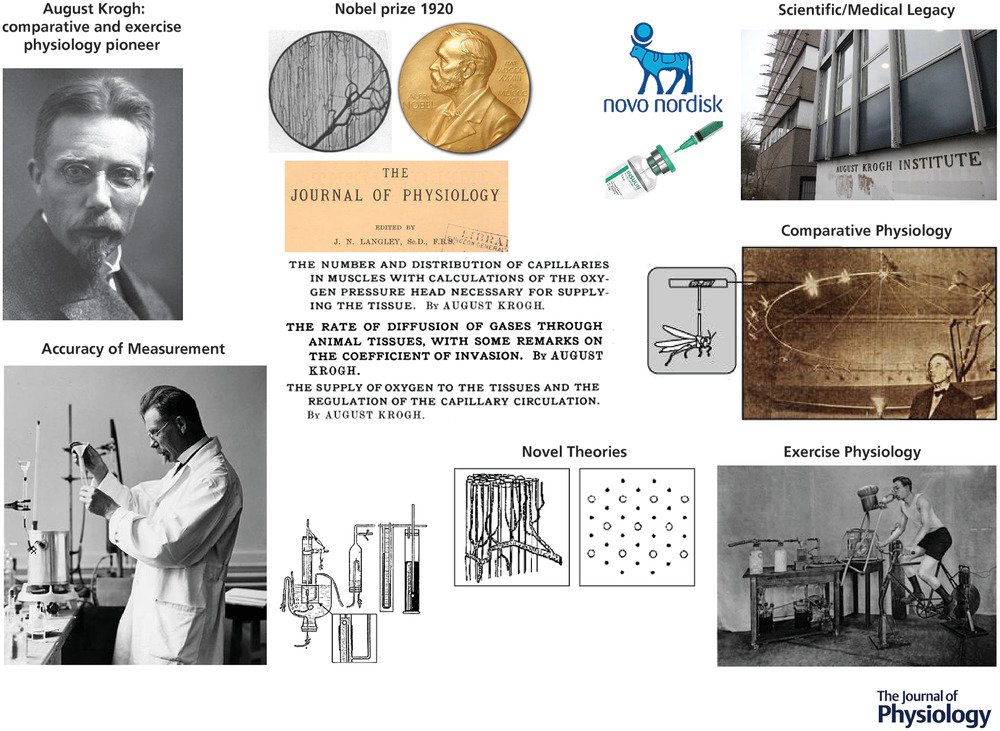
图1. 从毛细血管到肌细胞的PO2分布:克罗格理论与当代测量值
左图:根据克罗格(1919年a)的图6重新绘制,显示了由一根毛细血管(较小的内圆,半径 = r)供应的肌肉组织 “克罗格圆柱体”(半径 = R)。克罗格 - 厄兰方程(文中的方程(1)和(2))用于计算从毛细血管到组织中任何一点(例如上面的x点)的氧气分压降。在方程(1)中设x = R,得到组织圆柱体的最大半径,此时氧气分压>0 mmHg。从休息到收缩的克罗格式毛细血管募集模型减小了R,但没有考虑到随着代谢需求的增加,毛细血管内、间质或肌细胞内扩散能力(VO2)的变化。顶部 = 休息,底部 = 收缩。请注意,随着更多毛细血管被募集,R将减小,并且如果没有随着收缩而减小R(即毛细血管募集),将会有大量的缺氧组织体积(左图中的蓝色圆圈,中图中的蓝色箭头)。中图:克罗格 - 厄兰方程预测的氧气分压分布。右图:来自贝恩克等人(2001年,2003年)、麦克多诺等人(2005年)、平井等人(2018年)的测量值(大鼠慢肌和快肌以及混合肌肉,磷光猝灭法,微血管和间质氧气分压)、霍尼格等人(1997年)(狗股薄肌,低温显微分光光度法,肌细胞内PO2)、理查森等人(1995年,2006年)(人类股四头肌,P - MRS,肌细胞内PO2)。
在这里,我们还将研究导致克罗格提出毛细血管募集假设的主要事实,并尽可能在当时可用的技术和信息的背景下看待克罗格在1919年呈现的数据。然后,我们将根据后来的研究和当今可用的最先进技术回顾性地重新评估克罗格的发现和想法,以记录进展并使读者了解该领域的最新情况。将会变得明显的是,虽然他的观察、想法和理论极大地推动了该领域的发展,但对他的一些主要概念的严格坚持导致了新的观察和证据被要么一概而论地驳回,要么在很大程度上被忽视。
以下部分基于克罗格1919年的论文(克罗格,1919年a、b、c)及其中的相关引述,以帮助读者快速了解支撑毛细血管募集理论的观察和数据的逻辑发展。在某些情况下,我们参考了克罗格和他的女儿博迪尔以及其他人后来的出版物,这些出版物对于解释早期的发现和逻辑至关重要。其中最重要的是他的书籍,包括《毛细血管的解剖学和生理学》(克罗格,1922年)和《奥古斯特和玛丽·克罗格:科学人生》(施密特 - 尼尔森,1995年)。随后,从诺贝尔论文(及相关出版物)中得出的观察结果和前提将克罗格的发现置于当代的视角中。在相关的情况下,我们讨论了关键的方法学缺陷,包括当时和随后一个世纪的科学探究中存在的确认偏差。
如今,大量令人信服的实验证据使我们对毛细血管功能和血液 - 肌细胞交换的理解又回到了原点——尽管我们拥有了更多的机制性知识,特别是关于血液 - 肌肉氧气通量的动态控制、肌肉氧气分压以及休息和运动时骨骼肌之间和内部氧气需求与供应的精确匹配。具体来说,就像在克罗格(1919年a、b、c)之前一样,如今已知肌肉血流控制和血管阻力的主要部位位于毛细血管上游的动脉/小动脉内,而不是在毛细血管本身(综述:杜林和多拉,1997年;劳克林等人,2012年)。肌肉罗盖特细胞(现在已知为周细胞)的收缩性曾被认为是毛细血管募集的 “驱动力”,但这仍然存在争议,而且在过去的一个世纪里,没有确凿的证据表明健康的骨骼肌中的毛细血管能够在肌肉收缩时自行关闭血流或主动扩张以启动血流。
作为克罗格今天的一项伟大遗产,微循环领域正在蓬勃发展,肌肉氧气扩散能力()与灌注氧气传导率()一起,仍然是氧气输送的关键决定因素。现在人们认识到,氧气扩散能力不仅仅由先前不流动的毛细血管的募集来控制。相反,从休息到运动时,毛细血管红细胞通量和血细胞比容的增加以及已经有血液流动的毛细血管内毛细血管表面积的更大纵向募集,与增强的肌细胞内氧气运输能力一起,共同作用以增加氧气扩散能力和血液 - 肌细胞氧气通量(图2A;费德斯皮尔和波普尔,1986年;格罗贝和瑟夫斯,1990年;罗卡等人,1992年;综述:普尔,2019年;安格利斯和厄斯特高,2020年)。
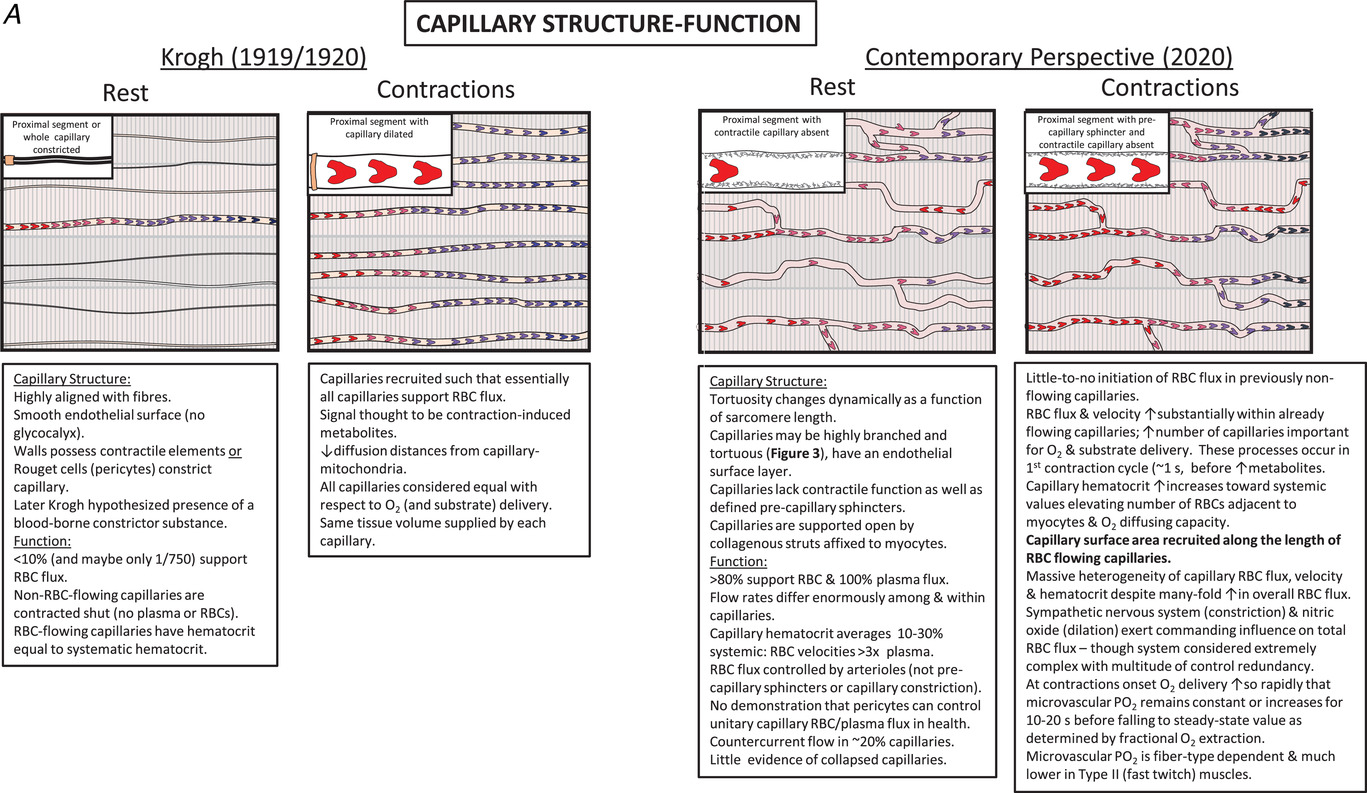
图2
A,从1920年到今天,科学发现彻底改变了我们对骨骼肌毛细血管结构和功能的理解(改编自普尔等人,2013年)。B,1919/1920年(左)和2020年当代视角(右)下休息和收缩时肌细胞结构和功能的关键要素总结。请注意,与以前认为的明显分开的 “豆形” 线粒体(左图插图)相比,右图插图中是复杂的相互连接的线粒体网络。请参阅支持信息视频S1,了解健康大鼠肌肉中的毛细血管红细胞血流动力学以及心力衰竭的影响。
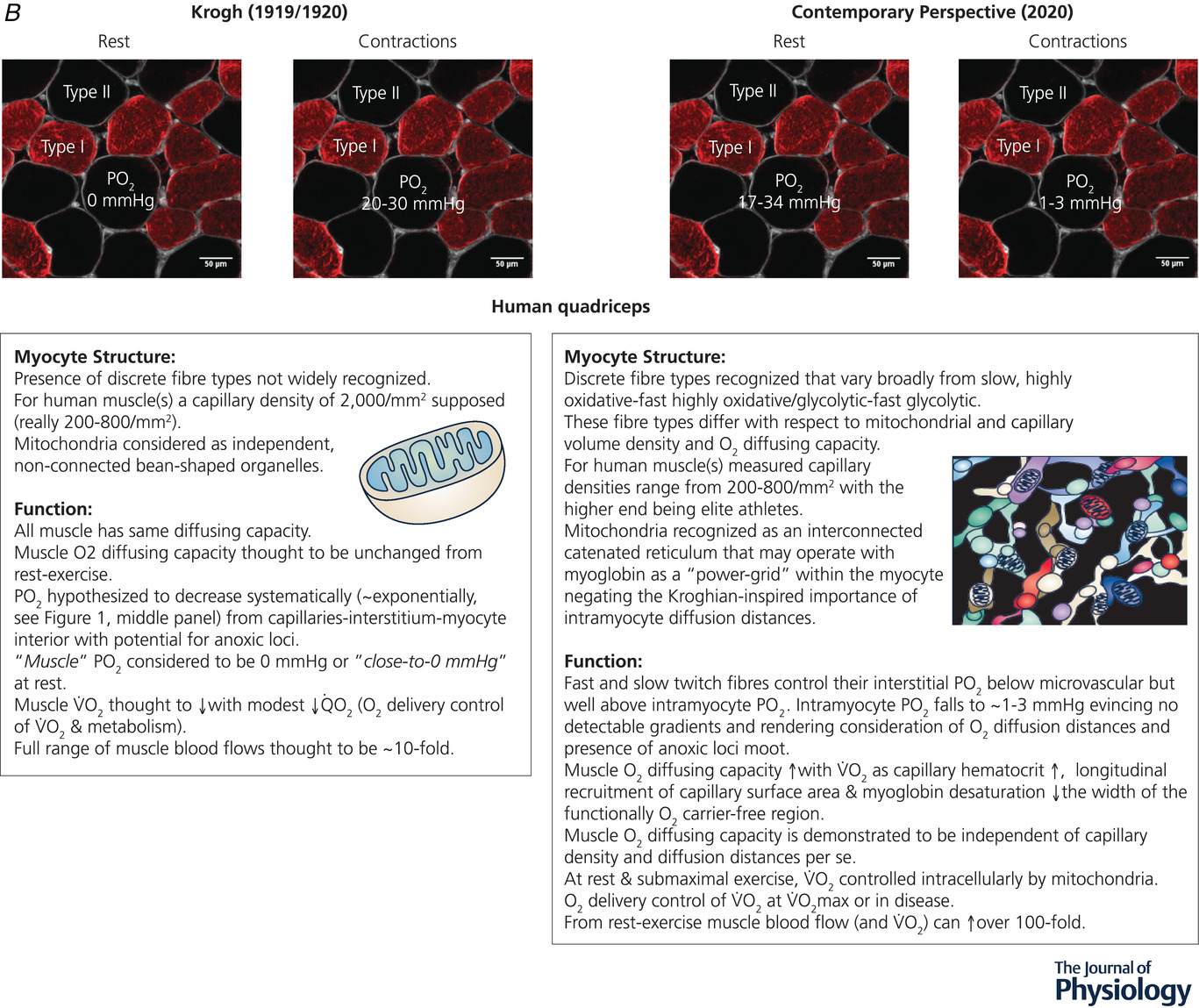
(图2续)
A,从1920年到今天,科学发现彻底改变了我们对骨骼肌毛细血管结构和功能的理解(改编自普尔等人,2013年)。B,1919/1920年(左)和2020年当代视角(右)下休息和收缩时肌细胞结构和功能的关键要素总结。请注意,与以前认为的明显分开的 “豆形” 线粒体(左图插图)相比,右图插图中是复杂的相互连接的线粒体网络。请参阅支持信息视频S1,了解健康大鼠肌肉中的毛细血管红细胞血流动力学以及心力衰竭的影响。
以免读者认为100年前的论文已经过时,或者克罗格的毛细血管募集假设对微循环控制和肌肉氧合的进展不再有影响,请参考以下文章:关于辩论,请参阅普尔等人(2008年a、b)和克拉克等人(2008年a、b);普尔(2014年a、b)和巴雷特等人(2014年);麦克拉奇等人(2020年)和凯斯克等人(2020年);关于综述,请参阅普尔等人(2013年);普尔(2019年);安格利斯和厄斯特高(2020年)。
克罗格的毛细血管募集假设
克罗格的毛细血管募集假设是在他试图将实验结果与他的毛细血管圆柱体模型相协调时提出的。
“假设每根毛细血管都独立于其他毛细血管向其周围的圆柱体组织供应氧气,这样不会犯严重的错误。”(克罗格,1919年a,第410页)
克罗格通过克罗格 - 厄兰方程(由数学家阿格纳·K·厄兰开发)将这些独立的圆柱体付诸实践,该方程预测了此类圆柱体中任何位置的组织氧分压(PO2):
(1)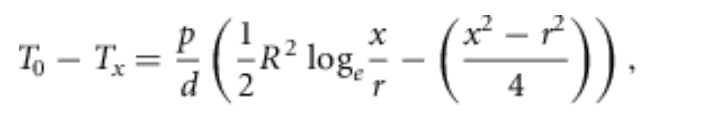
其中T0和Tx分别是毛细血管中以及距离毛细血管x处组织中的氧气分压值,R是组织圆柱体的半径,r是毛细血管半径,p是氧气消耗量(),d是氧气的扩散系数乘以其溶解度。
设x = R并将自然对数替换为常用对数,这个表达式预测了为肌肉供应氧气所需的氧气分压差:。
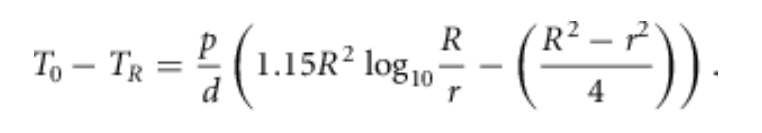
(2)
因此,若T₀ - T_R(毛细血管与组织圆柱体边界的氧分压差)小于静脉氧分压,那么“肌肉内所有区域的氧分压都将为正值”。但如果该差值超过静脉氧分压,就会出现氧缺乏,且在毛细血管静脉端附近尤为明显。
克罗格尝试用该方程估算R值(R为流动毛细血管间平均距离的1/2),进而推算肌肉组织内的毛细血管密度。克罗格假设,相邻肌肉圆柱体交界处的氧分压T_R可忽略不计(即接近0 mmHg),这一假设的依据来自费扎尔(Verzar)的研究——费扎尔曾在剑桥大学巴克罗格夫特实验室研究犬类肌肉,并发现肌肉氧分压会随氧气供应减少而降低(Verzár, 1912)。克罗格对这一发现的解读如下:
“……对于肌肉,费扎尔(1912)发现其(静息状态下的)氧分压必定相对较低,甚至可能为零。”
此外,克罗格实验室的高德尔(Gaarder, 1918)发现,当将水中氧分压提升至760 mmHg以增加氧气可利用度时,鱼类的氧分压会随之升高。基于这一结果,克罗格得出结论:“氧气从毛细血管向组织的扩散无法维持正值的氧分压”(Krogh, 1919c, 第458页)。
通过自制的自行车测力计,克罗格与林哈德(Krogh & Lindhard, 1913, 1915)发现,当肌肉耗氧量(p)增加10倍时,混合静脉血的氧分压会从约35 mmHg降至12 mmHg。他据此推测:“……在高强度运动时,静脉血的氧饱和度百分比几乎可以代表工作肌肉内的氧分压状况”。因此,当克罗格将方程(2)应用于运动和静息状态时,在假设(开放)毛细血管数量(R)和氧气扩散系数与溶解度乘积(d)保持恒定的前提下,他发现了一个矛盾:随着耗氧量(p)增加10倍,方程(2)的右侧值也会同步增加——但与此同时,氧分压驱动压力(方程左侧,且T_R≈0)却反而降低。
克罗格排除了“毛细血管半径大幅增加可解释该矛盾”的可能性:
“血液流经毛细血管的速率增加,甚至因血压升高导致的毛细血管被动扩张,显然都无法解释这一矛盾。”(Krogh, 1919c, 第458-459页)
这一结论促使克罗格推测:“……唯一可能的补偿机制是改变R值(即毛细血管与组织的距离),因此可以推断,肌肉内毛细血管间的平均距离应与气体交换呈反比变化……”(Krogh, 1919a)。
这种推理,再加上他“毛细血管周围距离R处的氧分压降至0”的观点,让克罗格坚信:毛细血管的开放比例(以及由此带来的流动红细胞毛细血管密度)必须与肌肉耗氧量(方程1和2中的p)成正比,即:“……每平方毫米横截面上的毛细血管数量应与气体交换呈正比”(Krogh, 1919a, 第412页)。
从现代视角审视克罗格-厄兰方程与毛细血管募集的七大基础假设
假设一:毛细血管总量估算
克罗格估算人体骨骼肌的毛细血管总长度约为10万公里(Krogh, 1922, 第10页)。如图2B所示(图3也隐含此信息),这一数值存在严重高估——若要灌注所有这些毛细血管,所需的血量将远超人体总血量。这一考量可能是克罗格坚信“多数毛细血管在静息时处于关闭状态”的关键原因。进而,这一假设强化了他的另一个观点:必须存在一种强效机制,能在非代谢需求时阻止毛细血管流动;基于此,他提出“体液性毛细血管收缩物质”的假说。他认为这种物质可能是“垂体后叶激素(pituitin)”(Krogh, 1922),其作用机制为:在收缩的毛细血管内,该物质会随时间逐渐分解,从而使毛细血管周期性开放,红细胞通量也随之周期性恢复。
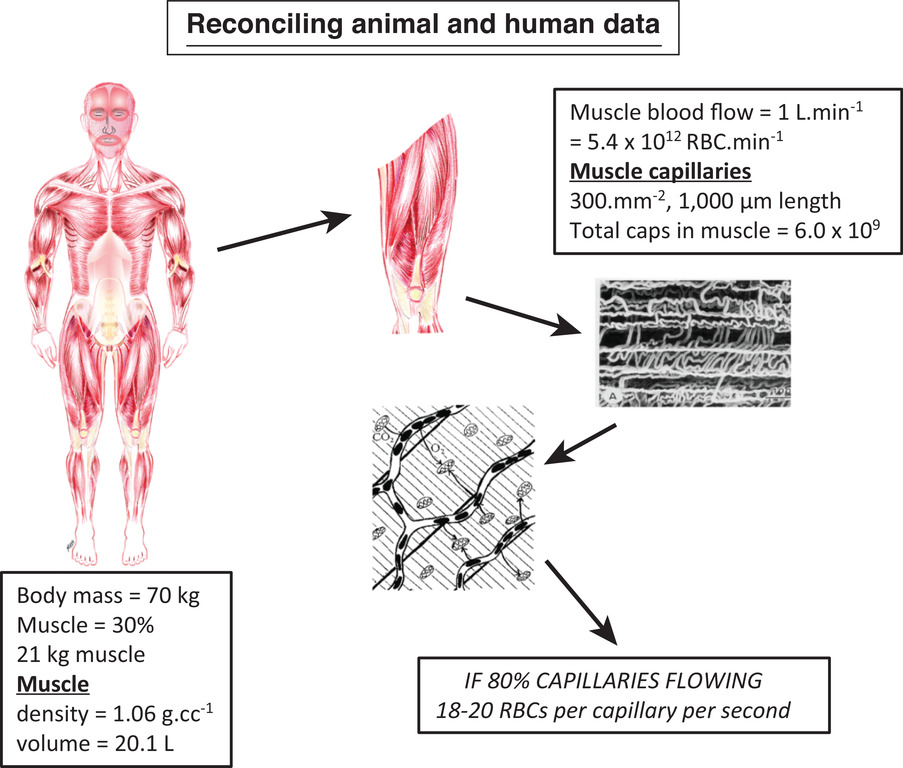
图3 动物体内测得的毛细血管血流动力学对人类是否“适用”?
活体显微镜观察(如Kindig等人, 1999, 2002)及染料灌注实验(Kayar & Banchero, 1985; Snyder等人, 1992)在麻醉和清醒动物中的结果均支持“静息肌肉中多数毛细血管存在红细胞通量”的观点。这一结论是否也适用于人类?本图结合已确定的肌肉血流量数据(如Rowell, 1974, 1993)与最佳实践测得的肌肉毛细血管密度估算值(如Hoppeler等人, 1985; Richardson等人, 1994; Sarelius, 1990)显示:理论上,人类肌肉中约80%的毛细血管可维持每秒18-20个红细胞的通量,这与大鼠斜方肌中测得的数值(每秒每毛细血管15-20个红细胞;Kindig等人, 1999, 2002)接近。与最左侧和中间方框不同,最右侧方框中斜体大写字母提示读者:“80%”这一数值取自动物研究文献,目前尚无人类相关数据。插图由医学插画师Mal Rooks Hoover提供。详见正文(改编自Poole, 2019)。
现代视角
克罗格假设毛细血管密度为2000个/平方毫米(Krogh, 1922, 第10页),但后续对人类股外侧肌的测量显示,更实际的数值为200-500个/平方毫米(至少在非精英运动员中如此)(Saltin & Gollnick, 1983; Hoppeler等人, 1985; Richardson等人, 1994)。此外,克罗格假设研究对象拥有50公斤肌肉,而对于体重70公斤的男性,肌肉量的合理平均值应为体重的30%,即21公斤。由此计算,若要灌注10万公里的毛细血管,在血细胞比容为45%的情况下,需13.2升血量——这显然远超人体总血量!
基于实测值的更合理估算显示,人体骨骼肌毛细血管总长度约为6000-15000公里;在平均血细胞比容为15%的情况下,仅需约183-460毫升血液(即占人体总血量的3%-8%)即可灌注。直到20世纪70年代,布莱恩·杜林(Brian Duling)及其同事在啮齿类动物肌肉中开展研究后,才证实哺乳动物微血管的血细胞比容远低于全身水平,随后这一结论在多个实验室中得到验证(Klitzman & Duling, 1979; Klitzman等人, 1982; Sarelius & Duling, 1982; Desjardins & Duling, 1990; Kindig等人, 2002; 综述:Secomb等人, 1998)。若克罗格当时意识到肌肉毛细血管总容积实际如此之低,就不会认为“静息肌肉中毛细血管需要收缩/关闭”了。
值得注意的是,运动训练带来的最早适应性变化之一是血容量增加:初期通过血浆量增加实现,随后通过造血作用使全血量和总血红蛋白量上升(Saltin & Gollnick, 1983; Schmidt等人, 1988; Green等人, 1991; Stevenson等人, 1994)。这些变化通常先于或伴随训练诱导的毛细血管新生。因此,对于运动肌肉毛细血管密度可达800个/平方毫米的精英运动员(Saltin & Gollnick, 1983; Hoppeler等人, 1985; 综述:Richardson等人, 1994)而言,新增的毛细血管容积在其总血量中所占比例可能并未增加——尤其是因为毛细血管密度的提升主要局限于受训肌肉。
克罗格圆柱体模型与克罗格-厄兰方程
如今我们已知,影响灌注性(QO2)和扩散性(DO2)氧通量的因素远多于克罗格当时的考量。
首先,灌注性氧通量主要由“收缩肌纤维旁毛细血管中的红细胞数量”决定(图4,中;Federspiel & Popel, 1986; Groebe & Thews, 1990;另见Malvin & Wood, 1992; Hirai等人, 2015, 2018)。静息肌肉中,不同毛细血管的血细胞比容存在显著异质性,平均值约为15%。当肌肉收缩时,红细胞通量和流速增加,灌注性氧通量会随以下因素动态上升:毛细血管血细胞比容升高、红细胞间距缩小、氧气释放分数增加——这些因素共同促进毛细血管表面积的纵向募集。
因此,不能将毛细血管视为“氧气输送与交换潜力相同且固定的单位”(另见Poole, 2019)。相反,即使在持续有红细胞流动的毛细血管内部及之间,毛细血管的几何形态、血细胞比容、红细胞通量和流速也存在巨大异质性(综述:Ellis等人, 1983; Groom等人, 1984; Koga等人, 2007; Fraser等人, 2012a,b, 2013; Koga等人, 2014; Heinonen等人, 2015; Poole, 2019)。这一认知对“单根毛细血管独立向特定组织区域供氧”的核心假设提出了挑战。卡瓦略与皮特曼(Carvalho & Pittman, 2008)在仓鼠颊囊牵缩肌中发现了纵向和径向的氧分压梯度,且氧气会从毛细血管扩散至更大的血管(Pittman, 1995, 2011; Golub & Pittman, 2013)。
更复杂的数学模型已通过数字模拟考虑了毛细血管网络间的扩散相互作用(Grunewald & Sowa, 1977),并开发了解析方法,以更好地理解“具有不同红细胞流速、入口氧分压和氧气结合特性的毛细血管层”之间的相互作用(Popel, 1980, 1982; Liu等人, 2012; Fraser等人, 2012a,b, 2013)。此外,多数动物和人类肌肉由混合肌纤维类型构成,肌纤维大小、耗氧量和肌纤维类型特性的差异会导致显著的氧分压空间异质性(Saltin & Gollnick, 1983; Liu等人, 2012)。如下文(假设六:现代视角及支持信息视频S1)所示,毛细血管间血细胞比容的差异对决定特定毛细血管的瞬时氧分压至关重要。
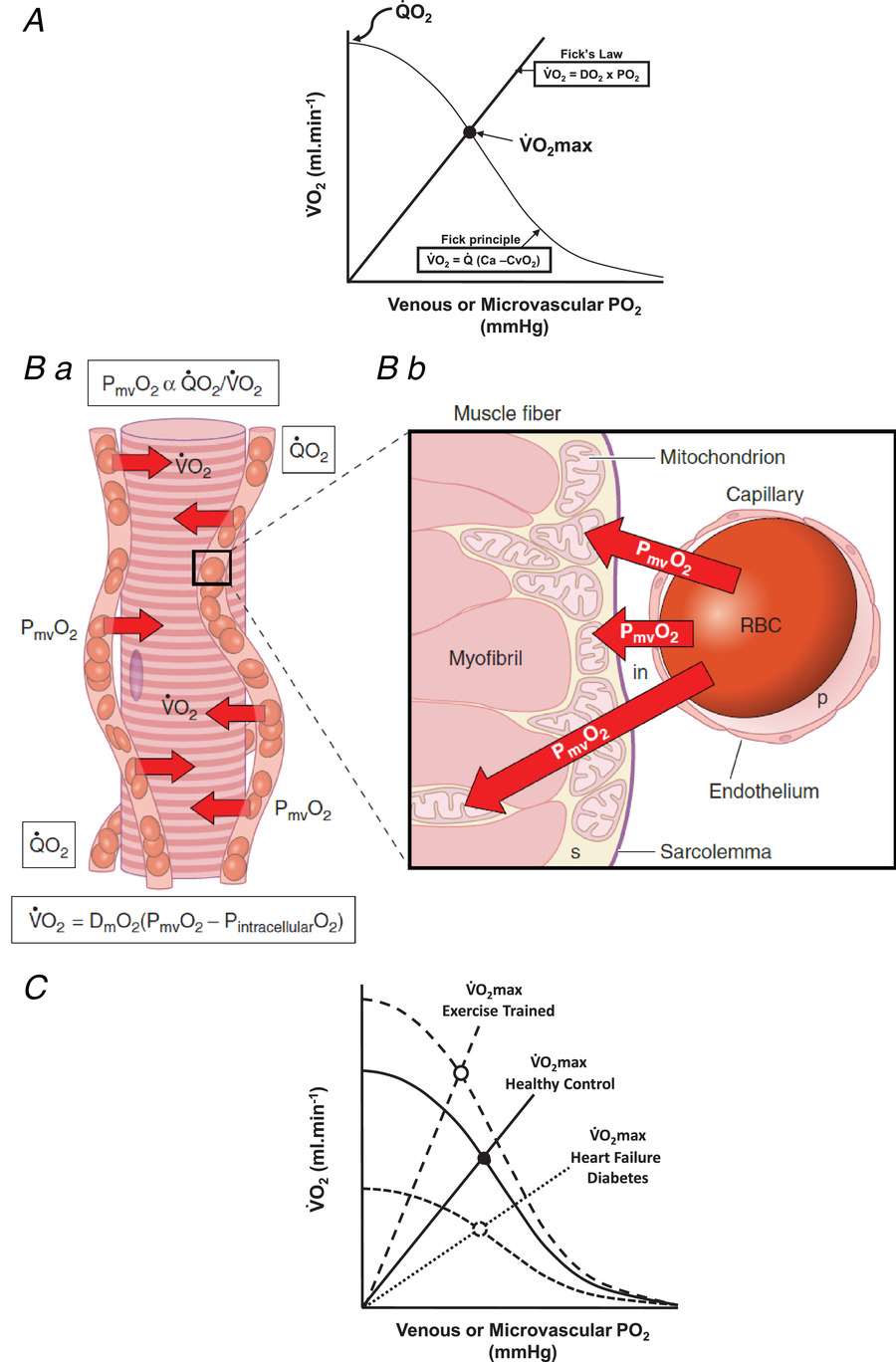
图4 灌注性与扩散性氧传导共同实现氧气摄取
A 图
该图最初由彼得・瓦格纳(Peter Wagner)在罗卡等人(Roca et al., 1992)的研究中提出,现以其名字命名为 “瓦格纳图”。将菲克定律(从原点出发的直线,V˙O2=DO2×ΔPO2)与菲克原理(V˙O2=Q×(CaO2−CvO2))相结合,可清晰理解 “共同构成特定最大摄氧量(即V˙O2max)的灌注性氧传导与扩散性氧传导”。其中:
V˙O2 = 摄氧量
DO2 = 肌肉或全身的氧扩散能力
Q = 血流量或心输出量
CaO2、CvO2 = 分别为动脉血氧含量与静脉血氧含量
B 图(a、b)
展示毛细血管与骨骼肌纤维间 “灌注性氧传导(GO2)” 与 “扩散性氧传导(DO2)” 的结构及功能决定因素。微血管中的氧分压(PO2)由V˙O2/Q比值决定。根据菲克扩散定律,任一时刻的摄氧量(V˙O2)必定是 “跨膜氧梯度(PO2cap−PO2im)” 与 “氧扩散能力(DO2)” 的乘积(V˙O2=DO2×(PO2cap−PO2im))。
由于肌肉收缩时,肌细胞内氧分压(PO2im)极低(约 1-5 mmHg,见图 2B),因此微血管氧分压(PO2cap)可近似视为驱动跨膜氧通量的压力(即PO2cap≈ΔPO2)。相应地,灌注性氧传导(GO2)是该氧分压(PO2cap)与血流量(Q)的乘积,而血流量本身主要取决于 “肌纤维旁流动毛细血管中的红细胞数量”。
如图 C 所示,运动训练可通过两种方式提升摄氧量(V˙O2):一是通过增加心输出量与肌肉血流量(Q)来提高灌注性氧传导(GO2);二是通过新增毛细血管(每条毛细血管均维持自身血细胞比容,并助力灌注性氧传导升高)来提高扩散性氧传导(DO2)。与之相反,心力衰竭、糖尿病等疾病会通过减少流动毛细血管数量,同时损害灌注性氧传导(GO2)与扩散性氧传导(DO2)(综述:Poole et al., 2012; Hirai et al., 2015)。(图改编自 Hirai et al., 2015)
C 图
瓦格纳图展示了两个核心过程:一是运动训练如何通过改善 “灌注性氧通量(Q与GO2)” 与 “扩散性氧通量(DO2)”,共同提升摄氧量(V˙O2);二是心力衰竭(Richardson et al., 2003; Poole et al., 2012; Hirai et al., 2015)、糖尿病(Padilla et al., 2006)等疾病如何导致上述氧通量下降,进而降低摄氧量(V˙O2)。有关心力衰竭对毛细血管红细胞通量的影响,详见支持视频 S1。
血细胞比容、红细胞通量与流速的异质性
如今我们认为,血细胞比容、红细胞通量与流速的异质性源于微循环的生物物理特性及内皮表面层,而非毛细血管直径的主动调节。目前已知,毛细血管内皮细胞表面覆盖着一层模糊的糖蛋白 / 透明质酸层,有时被称为 “糖萼(glycocalyx,意为‘甜蜜的外壳’)”,其向管腔内部延伸约 0.3 微米(Desjardins & Duling, 1990; Brown et al., 1996; Vink & Duling, 1996; Nieuwdorp et al., 2008)。
该表面层会减缓血浆平均流速,但允许红细胞在其表面轻松且快速滑动,从而有效降低毛细血管血细胞比容。在化学物质诱导或收缩诱导的血管舒张过程中,升高的红细胞通量与流速会使毛细血管血细胞比容向全身水平靠拢(可能通过影响内皮表面层实现),进而提升灌注性氧传导(GO2)(综述:Poole, 2019)。
克罗格本人曾注意到微循环中存在 “血浆撇取(plasma skimming,又称相分离)” 现象,并指出 “这一现象可能是毛细血管血液红细胞计数出现诸多异常的原因”(Krogh, 1921)。如今已明确,小动脉及小动脉 - 毛细血管分叉处会发生显著的血浆撇取;这种被称为 “法勒效应(Fahreaus effect)” 的现象,与内皮表面层共同作用,导致静息状态下肌肉毛细血管的血细胞比容降至全身水平的约 30%(即绝对数值约 15%)。而在肌肉收缩或其他充血状态下,毛细血管血细胞比容会向全身水平升高(Klitzman & Duling, 1979; Klitzman et al., 1982; Sarelius & Duling, 1982; Desjardins & Duling, 1990; Kindig et al., 2002; 综述:Secomb et al., 1998)。
非收缩性顺应性微血管网络
在非收缩性的顺应性微血管网络中,微血管流速与红细胞转运时间的标准差,往往与其在毛细血管床中的平均值呈比例关系(Rasmussen et al., 2015)。因此,静息状态下毛细血管红细胞流速的异质性,以及血液供应增加时流速的均一化,很可能由 “决定分支点血流动力学的生物物理特性” 与 “糖萼相互作用” 共同决定。
值得注意的是,当克罗格 - 厄兰方程(Krogh-Erlang equation)应用于 “表现出流速均一化与血细胞比容升高的开放毛细血管” 时,能够解释运动中观察到的 “血液 - 肌细胞氧通量增加约 80 倍” 这一现象(Angleys & Østergaard, 2020)。
克罗格圆柱体模型的其他简化问题
克罗格圆柱体模型在其他方面也存在过度简化。目前认为,肌细胞内的氧扩散能力(DO2im)会受肌红蛋白的存在及其氧饱和度的调节(综述:Honig et al., 1997)。肌红蛋白的作用类似 “接力桶队(bucket-brigade)”:部分脱氧的肌红蛋白(半饱和氧分压P50≈3−5 mmHg)比完全氧合的肌红蛋白更易携带氧气,因此能在低氧分压(PO2im)条件下提高肌细胞内氧扩散能力(DO2im)。
相应地,在犬类腓肠肌 - 跖肌复合体中开展的精细研究(通过运动训练和肢体固定调控毛细血管密度,进而改变氧扩散距离)表明,摄氧量(V˙O2)与毛细血管间扩散距离无关(Hepple et al., 2000)。因此,当克罗格得出结论:“根据(克罗格 - 厄兰)公式,唯一可能的补偿机制是改变R值(即毛细血管与远端组织圆柱体的边界)…… 因此可以推断,肌肉内毛细血管间的平均距离应与气体交换呈反比变化……”(Krogh, 1919a, 第 412 页)时,他在这一问题上的认知受到了 “后续被证实为错误的边界条件” 的限制 —— 具体而言,是他假设 “毛细血管内和肌细胞内的氧扩散能力固定不变” 这一错误前提。实际上,在肌细胞内氧分压(PO2im)较低(但绝非零)时,肌细胞内的氧扩散能力(DO2im)会显著增强。
肌肉收缩时,平均毛细血管(即微血管)氧分压(PO2cap)会下降,这与动静脉氧分压差扩大(即氧提取分数升高)的趋势一致(Behnke et al., 2001, 2002, 2003; McDonough et al., 2005; Ferreira et al., 2006a; 综述:Poole et al., 2011, 2012, 2013; Hirai et al., 2018, 2019)。结合 “中高强度运动时肌细胞内氧分压(PO2im)保持稳定(约 1-5 mmHg;Richardson et al., 1995, 1999, 2006; Honig et al., 1997)” 这一事实可知:毛细血管氧扩散能力(DO2cap,随血细胞比容升高而增强)与肌细胞内氧扩散能力(DO2im)的提升,必定是运动中 “氧通量与肌肉摄氧量(V˙O2)增加” 的主要驱动因素 / 促进因素。
关于细胞内扩散距离
就细胞内扩散距离本身而言,肌红蛋白的转运功能(尤其是在 “富含肌红蛋白的氧化性肌肉” 中,当肌细胞内氧分压(PO2im)较低导致肌红蛋白部分脱饱和时),或许能缓解甚至显著改善氧扩散距离过远的问题(Honig et al., 1997; Clanton, 2019)。此外,埃尔斯沃思与皮特曼(Ellsworth & Pittman, 1984)在仓鼠中发现,氧化性肌肉(比目鱼肌)的氧扩散能力(DO2)是低氧化性肌肉(缝匠肌)的 2-3 倍,这一差异至少部分源于肌红蛋白的存在及其浓度差异。
在克罗格所处的时代,线粒体被认为是 “离散的豆状细胞器”—— 这是通过典型的肌肉纤维薄切片横向观察得出的结论。但如今我们已明确,线粒体形成了复杂的链状互联结构(图 2B 右侧插图),这种结构可能有助于将质子和 / 或氧气从肌膜下区域转运至肌细胞深处(Bakeeva et al., 1978; Glancy et al., 2014, 2015; Clanton et al., 2013; Clanton, 2019; Vincent et al., 2019)。上述发现均对 “扩散距离本身的重要性” 及 “肌细胞内氧扩散能力(DO2im)恒定” 这两个观点提出了挑战,而这两个观点正是克罗格毛细血管募集模型的核心基础。
“肌细胞内氧分压可忽略不计” 的假设
克罗格 - 厄兰方程的推导基于一个核心前提:肌肉内氧分压(PO2)随与最近毛细血管距离的增加而系统性下降,且在静息状态下,克罗格圆柱体边缘的氧分压必定为 0 mmHg(图 1 左图)。如前所述,这一观点源于两方面研究:一是费扎尔(Verzár)在犬类肌肉中开展的氧分压测量,二是克罗格实验室的高德尔(Gaarder)在鱼类中得出的研究结果。
后来,威尔斯(Wells, 1932)对高德尔的测量提出了质疑:在高德尔的实验中,鱼类被转移到新环境后,需要 12-24 小时才能稳定自身代谢。因此,氧分压(PO2)对氧气可利用度的依赖性,是由 “强加的实验条件” 导致的,而非 “生理调控本身”。下文的公式(3)与图 5 将更深入地探讨这一观点。
此外,费扎尔实验中犬类所使用的麻醉深度,很可能损害了平均动脉压,进而降低了肌肉血流量(Q),导致实验样本状态受损。从多个角度来看,“健康肌肉在静息或亚极量运动时存在氧供应限制” 这一观点已被彻底证伪。具体而言,静息状态下肌肉的氧提取分数通常 < 0.5,这意味着存在 “可进一步提取的氧储备”—— 若血流量(Q)下降,即可提取这部分氧气。因此,当血流量(Q)适度下降时,根据菲克原理,动静脉血氧含量差(CaO2−CvO2)的扩大可维持摄氧量(V˙O2)稳定,公式如下:
(3)
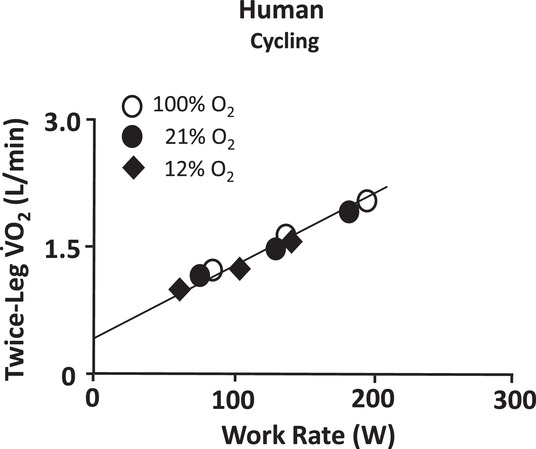
图 5 亚极量运动时的肌肉摄氧量不由动脉血氧含量决定
尽管骨骼肌在静息状态和亚极量运动时,可通过调节血流量来补偿动脉血氧含量的变化,进而调控向运动肌肉的氧气输送量(即氧气输送量),但值得注意的是,在人类中,肌肉摄氧量在很大程度上独立于吸入氧浓度和动脉血氧含量。数据来源于奈特等人(Knight et al., 1993)。犬类肌肉的相关佐证数据可参见布雷德尔等人(Bredle et al., 1989)的研究。
若不存在这种调节机制,心力衰竭(HF)患者在静息状态下的肌肉摄氧量会随心输出量同步下降,但实际情况显然并非如此(Abudiab et al., 2013);此外,血管扩张剂治疗及全身氧气输送量的增加,也不会提升这类患者在静息或亚极量运动时的肌肉摄氧量(Wilson & Ferraro, 1981; Chappell et al., 1983)。
目前已明确,由快肌纤维构成的肌肉与由慢肌纤维构成的肌肉相比,静息状态下的氧气提取分数更低,因此对氧气输送量降低的缓冲能力更弱(图 6 下图;Behnke et al., 2003; McDonough et al., 2005)。然而,即便亚极量运动可能会激活大量快肌纤维,在动脉血氧含量和 / 或氧气输送量适度降低时,稳态下的肌肉摄氧量仍保持不变 —— 至少在氧气输送量未低于某一临界低值前是如此。关于人类动脉血氧浓度升高或降低对肌肉摄氧量无影响的证据,可参见图 5(Knight et al., 1993);犬类肌肉的相关实例可参见布雷德尔及其同事(Bredle et al., 1989)的研究。
当前研究认为,在静息和亚极量运动的肌肉中,线粒体氧分压和 ATP 生成速率通过高能磷酸信号通路调控(Meyer & Foley, 1994; Poole & Jones, 2012)。因此,当氧气输送量高于某一较低临界值时(Bredle et al., 1989),即便氧气输送量下降,肌肉仍可通过以下方式维持摄氧量和 ATP 生成:提高氧气提取分数,以及代偿性增加细胞内游离 ADP 浓度、NADH + 浓度和 H + 浓度(Wilson et al., 1977; Arthur et al., 1992; Hogan et al., 1992),具体关系如公式(4)所示:
(4)
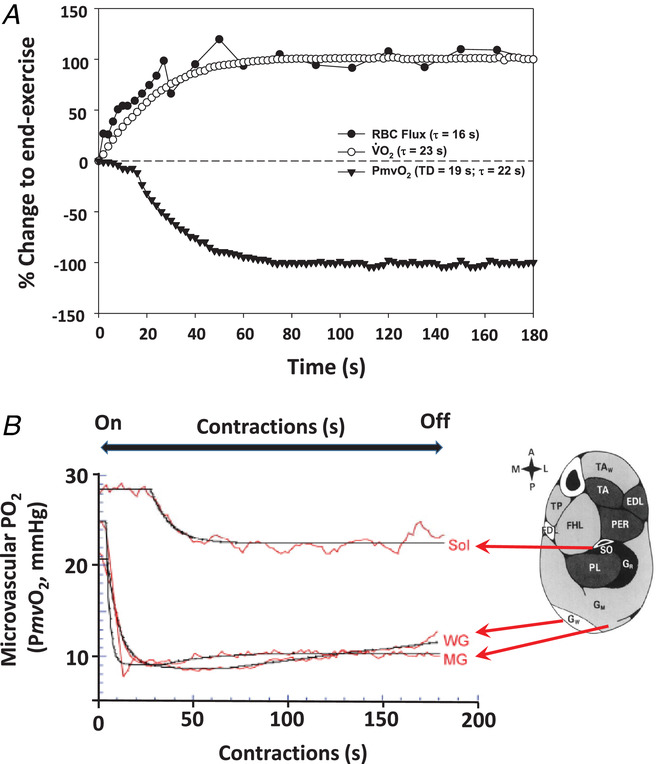
图 6 肌肉收缩开始后,毛细血管红细胞通量、肌肉氧气提取分数及摄氧量的快速动力学变化
任何关于毛细血管功能的假设模型,都必须具备足够快速的动力学响应,以匹配在肺部(综述参见 Poole & Jones, 2012)和收缩肌肉中观察到的生理行为(Behnke et al., 2001, 2002; Kindig et al., 2002; 综述参见 Poole, 2019; Poole et al., 2020a)。
A 图:在大鼠斜方肌(一种混合纤维型肌肉,氧化能力与人类股外侧肌相近)中,对 1Hz 电刺激诱导收缩(在时间 = 0 秒时开始)的响应显示:毛细血管红细胞通量(实心圆)和微血管氧分压(△PO₂,三角形)共同决定了肌肉摄氧量(V˙O2,空心圆),所有数据均标准化为响应幅度的 100%。数据来源于本克等人(Behnke et al., 2001)和金迪格等人(Kindig et al., 2002)。毛细血管红细胞通量通过高分辨率视频显微镜对单根毛细血管进行分析测得;微血管氧分压通过磷光猝灭技术测定(Rumsey et al., 1988; Poole et al., 1995; Behnke et al., 2001, 2002);肌肉摄氧量的估算方法为:结合测得的红细胞通量、动脉血氧含量和微血管氧分压(近似为毛细血管末端氧分压),并参考已建立的大鼠氧解离曲线(Altman & Dittmer, 1974)。图中显示了模型拟合结果:红细胞通量和微血管氧分压(而非肌肉摄氧量)均通过无延迟的单指数函数拟合。TD = 延迟时间,τ= 时间常数。该时间常数值与人类肌肉及肺摄氧量动力学第二阶段的时间常数相近(例如 Grassi et al., 1996)。图改编自普尔(Poole, 2019)。
B 图:通过磷光猝灭技术测得的微血管氧分压(△PO₂)变化曲线:记录了慢肌(比目鱼肌,Sol)和快肌(腓肠肌内侧头,MG;腓肠肌白肌,WG)从静息状态过渡到 1Hz 电刺激收缩状态的过程。微血管氧分压反映了肌肉微血管区内瞬时的氧气输送量(Q˙O2)与氧气消耗量(V˙O2)的比值。需注意:与慢肌相比,快肌的微血管氧分压下降更快、幅度更大,且最低值更低。数据来源于麦克多诺等人(McDonough et al., 2005)。这些结果,以及在大鼠中观察到的其他微血管氧分压数据(Behnke et al., 2003)和在人类股四头肌中通过近红外光谱(NIRS)获得的数据(基于脱氧血红蛋白 + 肌红蛋白浓度;Koga et al., 2015; Okushima et al., 2015)均表明:不同肌肉及不同肌纤维类型对 “氧气输送量 - 氧气消耗量”(进而对微血管氧分压)的调控存在差异。有趣的是,这一现象支持了以下观点:通过 “氧气输送量 - 氧气消耗量” 比值对肌肉血流量进行上游(即小动脉水平)调控,可能在肌肉收缩的代谢响应中发挥重要作用,并在一定程度上解释了运动训练和疾病状态下这种响应的变化(Poole et al., 2020a)。
微血管→组织间隙→肌细胞内的氧分压梯度降低,会导致细胞内游离 ADP 浓度升高,并刺激糖酵解。因此,即便肌细胞内氧分压下降未改变肌肉摄氧量,肌肉乳酸的生成和释放仍会增加(Richardson et al., 1998)。
此外,如图 2 下图所示,肌细胞内氧分压从静息状态到运动状态会急剧下降,而非如克罗格所认为的那样升高。
如今,一系列创新技术(尽管各有局限)揭示了与克罗格认知截然不同的肌肉氧分压分布特征(图 1、图 2、图 4、图 7),这些技术包括:肌肉微血管和组织间隙氧分压的磷光猝灭测量技术、质子磁共振波谱(P-MRS),以及静息和收缩 / 运动肌肉中肌细胞内氧分压的冷冻显微分光光度测定技术。具体而言,静息状态下(约 20-45 mmHg)和收缩状态下(约 6-25 mmHg)的微血管氧分压,受肌肉摄氧量、肌纤维类型以及氧化能力(可能)的调控(图 6 下图、图 8;Behnke et al., 2001, 2002, 2003; Geer et al., 2002; Hirai et al., 2018, 2019)。在从红细胞到组织间隙这一极短的物理空间内(即 < 1 微米),氧分压会显著下降约 8-10 mmHg,且这种下降在肌肉收缩过程中持续存在(图 8;Hirai et al., 2018, 2019; Craig et al., 2018; Poole, 2019)。
无论是静息还是收缩状态,均无证据表明肌细胞内氧分压会降至克罗格所认为的 0 mmHg—— 而这一假设正是其毛细血管募集理论的核心(见图 2B)。通过传统技术(氧电极:Whalen et al., 1974, 1976)和更先进的技术(快速冷冻犬肌肉中的肌红蛋白饱和度测定:Honig et al., 1997;质子磁共振波谱:Richardson et al., 1995, 1999, 2006)测得 / 计算出:动物和人类在静息状态下的肌细胞内氧分压为 17-34 mmHg;收缩状态下,肌细胞内氧分压会急剧下降至 1-5 mmHg,但绝不会降至 0 mmHg(图 8;Richardson et al., 1995; Voter & Gayeski, 1995; Honig et al., 1997)。此外,值得关注的是,即便在如此低的氧分压下,左移的肌红蛋白解离曲线仍能确保肌红蛋白保持较高的氧饱和度。
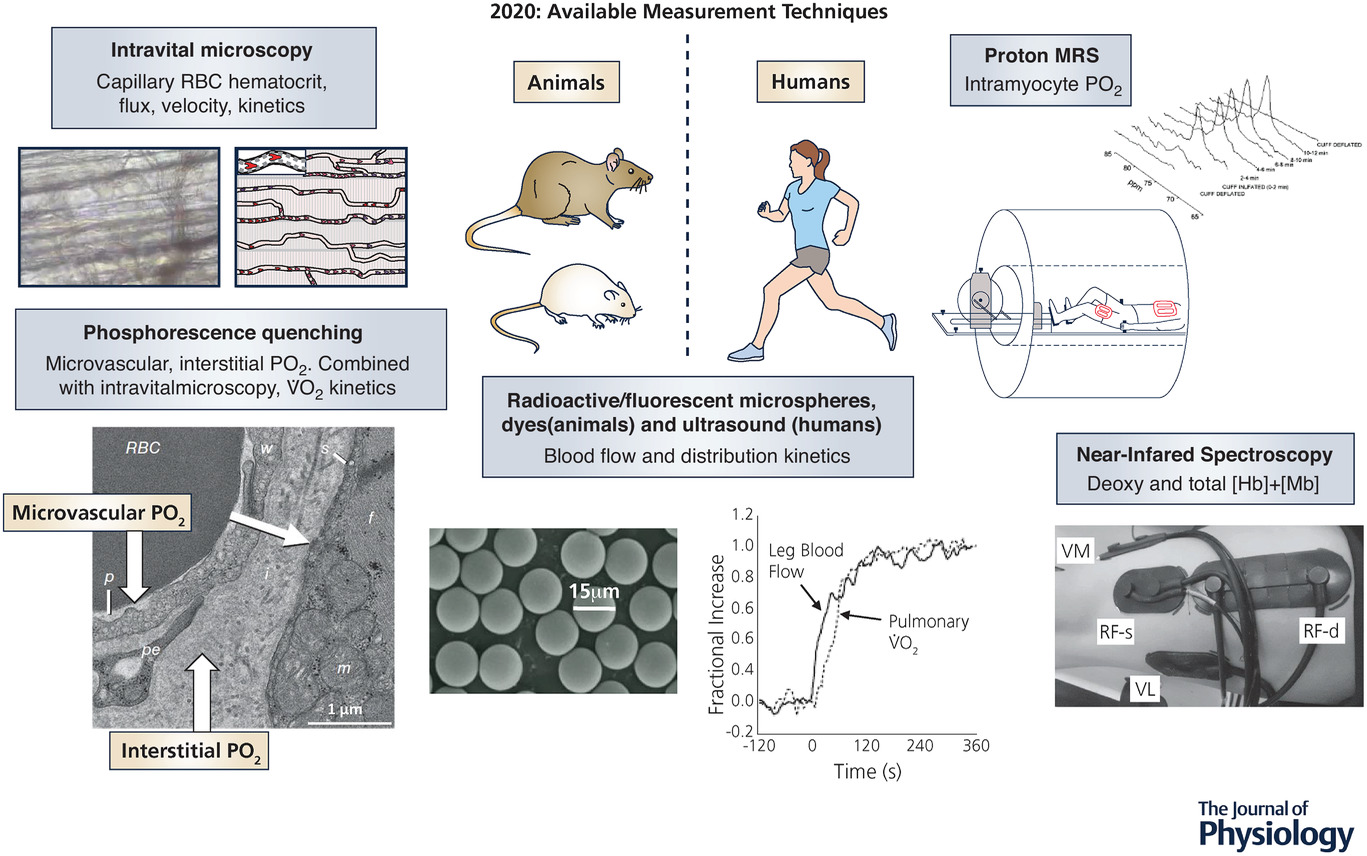
图 7 2020 年用于研究动物(左)和人类(右)微血管、组织间隙及肌细胞功能的技术
在这些技术中,只有活体显微镜(左上图)是克罗格在 1919/1920 年可使用的技术。从左上图开始按逆时针方向依次为:
活体显微镜技术(Kindig et al., 2002; Poole et al., 2013);
微血管和组织间隙氧分压测量技术(Behnke et al., 2001; Hirai et al., 2018);
用于肌肉 / 局部血流量分布测定的放射性标记微球技术(Musch et al., 2004);
肢体血流量的超声测定技术(Koga et al., 2007);
用于脱氧血红蛋白 + 肌红蛋白浓度及总浓度测定的近红外光谱技术(NIRS)(Koga et al., 2007, 2015; Lutjemeier et al., 2008; Davis & Barstow, 2013; Okushima et al., 2015, 2016);
用于肌细胞内氧分压测定的质子磁共振波谱技术(P-MRS)(Richardson et al., 1995, 2006)。
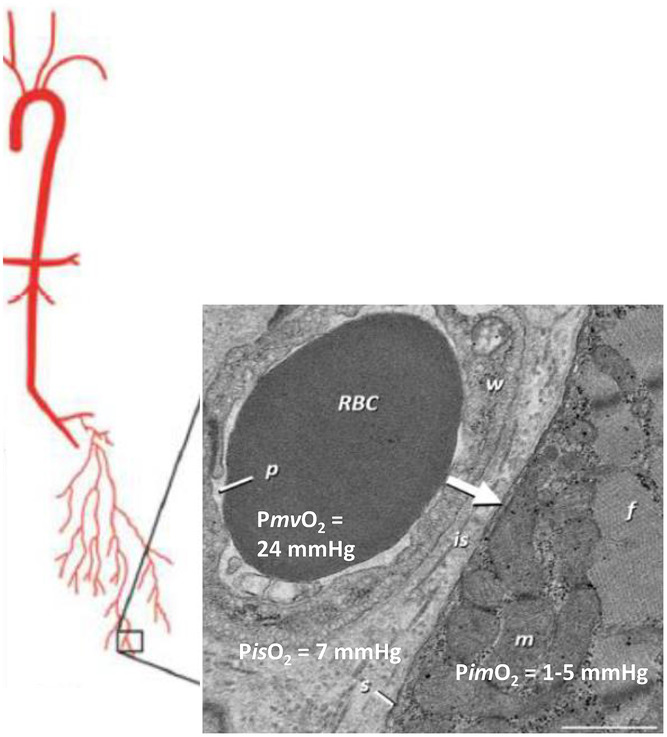
图 8 收缩状态下斜方肌的微血管氧分压和组织间隙氧分压(分别用△PO₂ₙᵥ和△PO₂ᵢₛ表示)
需注意从毛细血管(△PO₂ₙᵥ)到组织间隙(△PO₂ᵢₛ)、再到肌细胞内(△PO₂ᵢₘ)的显著跨膜氧分压梯度。图改编自平井等人(Hirai et al., 2018, 2019)。肌细胞内氧分压(△PO₂ᵢₘ)数据来源于理查森等人(Richardson et al., 1995)对人类股四头肌的研究。图中缩写:RBC = 红细胞,p = 血浆,w = 毛细血管内皮壁,is = 组织间隙,f = 肌原纤维,m = 线粒体网状结构。
克罗格认为,在极量运动和最大摄氧量状态下,随着更多(甚至全部)毛细血管开放,肌细胞内氧分压会显著高于静息状态(图 2B)。但对犬肌肉的冷冻显微分光光度分析(Honig et al., 1997)和对人类肌肉的质子磁共振波谱研究(Richardson et al., 1995, 2006)提供的直接证据表明,实际情况并非如此(图 1、图 2、图 7)。
克罗格对毛细血管募集假说的验证(1920年之前)
由于无法可靠测量活体肌肉中开放毛细血管的平均间距,克罗格向肌肉血管床灌注了他认为含有“超显微级”颗粒的印度墨汁(佩利坎珍珠墨水,冈瑟-瓦格纳公司生产)。假设克罗格使用的是小颗粒碳(而非约10微米的大颗粒碳;Ahlberg等人,1995),这类颗粒只有在聚集时才能被观察到,而聚集很可能会堵塞毛细血管内的血流(参见Stehbens & Florey,1960)。关键问题在于:静息肌肉血流量较低,相比运动/收缩时高血流量的情况,会有更多毛细血管被堵塞——无论实际是否存在毛细血管募集,这种现象都可能让人误以为存在募集过程。
克罗格计划对“多种不同动物”的肌肉进行注射,以验证他提出的“毛细血管密度与气体交换呈正比”的推论,但这一计划因“……难以实现肌肉组织的完全灌注……等意外困难”而受阻。他发现,通过以下方法可获得最佳结果:“……直接通过肌肉动脉注射,尽可能结扎所有动脉吻合支,并采用高压灌注”(Krogh,1919a,第412页)。而实现这一目标所需的灌注压力接近“一个标准大气压”!此外,他还发现,“……注射材料最好在动物处死后存放一两天再进行注射”。在注射过程中,他用普鲁士蓝对明胶进行染色,或使用“印度”(原文如此)墨汁,并在纵向和横向切片上计数毛细血管数量。
在探究代谢潜力与毛细血管密度的关系时,克罗格得出结论:“显然,仅凭如此少量的测定无法得出可靠结论……”,并对缺乏“可靠的灌注技术”感到遗憾(Krogh,1919a,第414页)。不过,通过结合鳕鱼、青蛙、马、狗和豚鼠肌肉的毛细血管密度计算值与代谢率,他得出:“……为肌肉供氧所需的氧分压梯度非常小……”,因此“……即便在代谢可能增加20倍的高强度肌肉运动中,肌肉组织的氧分压实际上仍将与静脉血氧分压相当……”,其中静息状态下T₀-T_R(即公式(2)中的氧分压差)范围为0.1-0.4 mmHg,而豚鼠在“高强度肌肉运动”时该值也仅达到约6 mmHg。
如今我们已知,与克罗格的结论“根据毛细血管总数推算出的必要氧分压梯度在所有情况下都极低”相反,肌肉收缩时,毛细血管-组织间隙-肌细胞内存在显著的氧分压差异(图1右侧;Hirai等人,2018,2019)。
假设二
“静息肌肉中的毛细血管循环变化很大,但通常血流微弱。这在一定程度上取决于麻醉状态。深度麻醉会导致大量毛细血管关闭,从而使刺激后产生的变化更显著”(Krogh,1919c)。
现代视角
克罗格向35-40克的青蛙体内注射了0.2-0.25克乌拉坦(氨基甲酸乙酯)——这一剂量是代谢率高出一个数量级以上的大鼠所用剂量的约5倍。根据他的报告,这些剂量似乎影响了实验动物的全身循环:躯干肌肉(如腹直肌)“血液充盈良好,几乎所有毛细血管中都能观察到血流”,而肢体肌肉中“有时很难发现开放的毛细血管”(Krogh,1919c,第461页)。在“经乌拉坦深度麻醉的青蛙中……舌头起初呈红色,但固定后逐渐变苍白,因为……仅能在少数乳突的毛细血管中观察到循环”。颏舌骨肌/舌骨舌肌也出现了相同情况。然而,几秒钟的强直收缩会导致“大量毛细血管显现”(Krogh,1919c,第460页)。
克罗格使用的乌拉坦及其他麻醉剂会损害心血管系统的反应能力,常导致血压下降,进而可能引发压力感受器介导的外周(包括骨骼肌)血管收缩(Armstrong等人,1982;Wixson等人,1987)。克罗格在实验中通常不会测量或报告麻醉对血压的影响,从他的表述(上文)中也无法明确:他是否将麻醉深度视为需要通过严格标准化实验条件来控制的误差来源(现代观点),还是将深度麻醉视为激发基础生理反应的手段。若为后者,克罗格在试图验证其理论计算时,很可能在无意中使实验产生偏差,导致自己倾向于观察到收缩过程中的毛细血管募集现象。
目前已明确,除麻醉外,多种其他实验条件也会影响毛细血管/微血管血流,进而可能降低骨骼肌中存在红细胞通量的毛细血管比例。其中最关键的条件包括肌肉拉伸、低血压、氧分压过高和创伤。
1. 肌肉拉伸:薄肌肉的光学特性更好,因此毛细血管的清晰度更高。这使得研究者容易在显微镜下拉伸目标肌肉。通常,静息状态下骨骼肌(心肌除外)的肌节长度约为2.6-2.7微米(Mathieu-Costello等人,1989;Poole等人,1997)。将肌节拉伸至3.0微米或更长会引发主动血管收缩,同时导致毛细血管管腔变窄,使大量毛细血管中的红细胞通量停止(Welsh & Segal,1996;Kindig & Poole,1999,2001)。而节律性收缩有望部分或完全逆转这种效应。
2. 低血压:除上述麻醉效应外,脱水、失血、吸入性缺氧或血管扩张剂导致的平均动脉压降低(尤其是降至70 mmHg以下),会诱发血管收缩,并在微血管氧分压降低的情况下减少存在血流的毛细血管比例(Behnke等人,2006)。详见本文末尾的支持视频S2。
3. 氧分压过高:高氧会导致小动脉收缩,因此必须确保实验动物吸入的氧气不会使动脉氧分压超过90-100 mmHg(即生理水平)。此外,暴露的肌肉必须通过保鲜膜等不透氧屏障与约150 mmHg(海平面环境)的环境氧分压隔离,或用经95%氮气/5%二氧化碳平衡(pH 7.4)的温热(约38°C)等渗溶液(如克雷布斯-亨塞尔特溶液)冲洗/灌注(Bailey等人,2000;Russell等人,2003;Ferreira等人,2006b;Padilla等人,2006;Copp等人,2009)。如Parthasarathi & Lipowsky(1999)的研究所示,将现有氧分压从接近生理水平的值(即35 mmHg)提升至150 mmHg的高氧水平,会通过小动脉(而非毛细血管)收缩导致毛细血管显著“去募集”。
4. 创伤:为进行显微镜观察而暴露肌肉时造成的手术创伤,会根据损伤部位和性质引发充血或缺血反应。同样,去除筋膜鞘等表层结缔组织(若必须进行)时必须极为小心,因为这也可能造成肌肉损伤。因此,研究者有责任证明其使用的活体肌肉标本未表现出非生理行为。例如,通过放射性标记微球测量发现,大鼠斜方肌活体标本的血流量与完整肌肉无差异(Bailey等人,2000)。
当采取预防措施排除这些干扰因素后,有充分的直接证据表明:大鼠斜方肌及其他肌肉中的绝大多数毛细血管都存在红细胞通量——尽管即使是来自同一终末小动脉的毛细血管,其红细胞通量、流速和血细胞比容也存在极大异质性(表1右侧;Poole等人,1997;Kindig & Poole,1998,1999,2001;Kindig等人,2002)。重要的是,在从静息到收缩的过渡过程中,该标本表现出以下特征:(1)血流量和摄氧量动力学与在体收缩肌肉(Hogan等人,1993;Grassi等人,2000;Clifford & Hellsten,2004;Clifford & Tschakovsky,2008)及人类自主运动(Knight等人,1993;Grassi等人,1996;Bangsbo等人,2000)中观察到的结果一致;(2)血流量与摄氧量的比值约为5-6:1,这是陆生非马科哺乳动物的典型特征(Knight等人,1993;Ferreira等人,2006a;Poole & Erickson,2011)。
表 1 两种对立的微循环观点 —— 按时间顺序排列的代表性文献
克罗格学派:原本 “关闭” 的毛细血管开放 现代观点:静息时多数毛细血管存在红细胞和 / 或血浆流
注:支持“静息时多数毛细血管存在红细胞和/或血浆流”的文献(右侧),多为对肌肉微循环的直接观察或采用毛细血管内皮染色的研究。右侧文献未使用1-甲基黄嘌呤(1-MX)或超声造影(CEU)等间接方法——这类方法的任何数据及其解读都依赖大量假设(左侧标*文献)。本表在Poole(2019)的基础上进行了扩展和更新,详见正文。
综上,采用先进技术,在定义明确且严格控制的条件下对温血动物和人类肌肉开展的现代研究(图2、图7),以以下证据反驳了毛细血管募集理论:
1. 通过活体显微镜观察发现,静息状态下多数肌肉毛细血管中存在血浆和红细胞通量(表1右侧)。
2. 目前尚无证据表明骨骼肌中存在毛细血管主动收缩或毛细血管前括约肌(Sakai & Hosoyamada,2013)。事实上,现已明确:骨骼肌和心肌中,毛细血管腔外表面与周围肌细胞之间的胶原支架可防止毛细血管塌陷(或收缩)(Caulfield & Borg,1979;Borg & Caulfield,1980)。
3. 从静息到运动状态下正常的血流量和摄氧量动力学,并不需要新的毛细血管募集(图6上图;Kindig等人,2002;Poole,2019)。这与克罗格在1910年发表的“七个小恶魔”系列论文中的论证大致相似——他在该系列论文中指出,波尔(Bohr)提出的“肺气血屏障存在主动氧分泌”的观点并无必要(Krogh,1910a-e;Krogh & Krogh,1910a、b)。现有研究结果表明,毛细血管募集并非骨骼肌收缩时满足自身氧需求的必要条件,因此迫切需要建立一个关于骨骼肌毛细血管功能的现代模型(Poole等人,2013;Poole,2019;Angleys & Østergaard,2020)。重要的是,凭借现有技术,无法证实或证伪静息或运动状态下人类所有骨骼肌毛细血管中是否存在红细胞通量(即是否存在“募集”),但关于动物肌肉的证据可参见下文第4点。
4. 在麻醉和清醒大鼠的静息状态下,几乎所有肌肉毛细血管中都观察到了血流(至少是血浆流)(Kayar & Banchero,1985;Snyder等人,1992)——这一观察结果排除了“新毛细血管募集”的可能性。值得注意的是,克罗格的直接观察大多是在深度麻醉的青蛙组织中进行的,而青蛙的红细胞大小和毛细血管几何形态与哺乳动物肌肉存在显著差异(图9)。此外,需注意青蛙是变温动物,其代谢率极低且依赖环境温度,同时适应在低氧环境中长时间存活。因此,在氧气供应和利用的关键方面,青蛙与哺乳动物存在巨大差异。
要不要我帮你整理一份文中关键实验方法(如印度墨汁灌注、活体显微镜观察等)的对比表?清晰列出每种方法的原理、克罗格时代的应用局限及现代改进,方便你快速理解技术发展对理论认知的影响。

图9 克罗格绘制的机械刺激前(上图)与刺激后(下图)青蛙舌头毛细血管图
该图未标注比例尺。青蛙的红细胞远大于人类或大鼠的红细胞。因此,无论这条血管能否被定义为毛细血管,其尺寸都异常粗大,直径可能约为50微米,这与哺乳动物骨骼肌毛细血管的结构(大小)或行为(显著主动扩张)均不相符。图源自克罗格的诺贝尔奖演讲(1920b)及克罗格(1922,图31,第124页)的著作。
(5)对动物和人类肌肉的已有测量显示,静息状态下的血流量足以让大部分毛细血管维持每秒18-20个红细胞的通量(图3;Poole等人,2000、2011、2013;Kindig等人,2002;Russell等人,2003;Hydren等人,2019;Poole,2019)。
(6)测量结果表明,静息状态下犬类(Honig等人,1997)和人类(Richardson等人,2006)肌肉的肌细胞内氧分压值显著较高(20-34 mmHg)(图1右侧面板、图2B)。
(7)大量近红外光谱(NIRS)研究显示,清醒人类在运动时,股四头肌中血红蛋白+肌红蛋白的总浓度仅比静息状态增加10%-30%(例如Davis & Barstow,2013;Okushima等人,2016;综述参见Poole,2019)。若静息时仅有不到10%的毛细血管(或如克罗格所假设的,豚鼠肌肉中仅1/750的毛细血管)含有红细胞,其余毛细血管在运动时才被募集,那么血红蛋白浓度应会增加9倍以上(或用克罗格的话说:“运动时,工作肌肉毛细血管中的血液量会大幅增加”),进而导致测得的血红蛋白+肌红蛋白浓度显著升高。但实际情况中,并未观察到如此显著的血容量增加。
(8)大量有力证据支持“肌肉血管调控位于小动脉水平”这一观点(综述参见Duling & Dora,1997;Laughlin等人,2012)。因此,小动脉的收缩/舒张与平均动脉压共同决定了以下因素:不仅包括哪些毛细血管存在红细胞通量,还包括通量的大小及毛细血管血细胞比容(Klitzman & Duling,1979;Klitzman等人,1982;Sarelius & Duling,1982;Desjardins & Duling,1990;Kindig等人,2002;综述参见Secomb等人,1998)。
假设三:扩散系数的测量(Krogh,1919b)
克罗格选择测量氧(VO2)在克罗格-厄兰方程中的常数d,测量对象涵盖青蛙腹壁(骨骼肌)、子宫(平滑肌)等不同“膜结构”。他在研究中提到:“因此,我怀疑我的实验中可能存在某种未知误差源。”随后,他重新设计了设备与实验方案,研究对象包括明胶和犬类腹膜(厚度8-12微米)。他明确证明:“氧的扩散速率随温度升高而增加(每升高1℃约增加1%)……”,同时发现肌肉的扩散常数(0℃、压差760 mmHg条件下为0.14 cm2·μm⁻1·min⁻1)略高于结缔组织,但氧在动物组织中的扩散速度远慢于在水或明胶中的扩散速度。
现代视角
克罗格所称的“扩散常数”,如今更常被称为“克罗格扩散系数”(K),其值等于特定分子在特定介质(如空气、肌肉)中的扩散系数(D)与该分子在该介质中的溶解度($alpha$)的乘积,即K = Dalpha$。克罗格测得的20℃条件下氧在“肌肉”(青蛙腹壁肌肉)中的“扩散常数”为0.14 cm3 O₂·(cm2·μm⁻1·min·atm)⁻1。将单位转换为更常规的度量标准(即将微米换算为厘米)可得:20℃时“肌肉”的K值为1.4×10⁻⁵ cm3 O₂·(cm·min·atm)⁻1;外推至37℃核心体温时,该值为1.85×10⁻⁵ cm3 O₂·(cm·min·atm)⁻1。
Kawashiro等人(1975)后续对大鼠腹壁的$K$值测量显示,37℃时该值为2.53×10⁻⁵ cm3 O₂·(cm·min·atm)⁻1。Bentley及其同事(1993)采用Mahler等人(1985)测得的青蛙缝匠肌氧溶解度值,计算得出37℃时青蛙缝匠肌的$K$值为4.28×10⁻⁵ cm3 O₂·(cm·min·atm)⁻1。Ellsworth & Pittman(1984)测得的仓鼠不同肌肉$K$值也比克罗格的结果高出约2倍,且高氧化型肌肉的扩散常数高于低氧化型肌肉。
扩散系数(D)与溶解度($alpha$)对温度的依赖性相反:$D$随温度升高而增大,$alpha$随温度升高而减小(Bentley & Pittman,1997;Bentley等人,1993;参见该文献表1)。由于$D$随温度的增大速率快于alpha的减小速率,因此$K$也随温度升高而增大(如克罗格所观察到的),但增大速率慢于D。因此,运动时肌肉温度可能从低于核心体温升至高于核心体温(例如35-40℃,Saltin等人,1968),而由于$D$呈指数级增大,氧在收缩肌肉中的扩散会更顺畅。
正如克罗格在“七个小恶魔”系列论文中对肺部氧扩散的错误结论(Gjedde,2010),他似乎未考虑到“无需毛细血管募集即可提高肌肉摄氧量(VO2)”的可能性。当时人们对肌红蛋白的了解极少,且他的许多观察对象是缺乏肌红蛋白的青蛙组织,因此并未考虑肌红蛋白的存在及其在促进肌细胞内氧运输中的作用——尤其是在收缩时肌红蛋白去饱和的情况下,肌红蛋白能显著提高氧扩散效率(参见Honig等人,1997)。
他在测量氧扩散时,为肌肉创造了高氧微环境(氧分压=760 mmHg),这很可能抵消了肌红蛋白对肌内氧扩散的促进作用(即功能性氧载体耗竭,Honig等人,1997)。此外,“静息肌肉的毛细血管血细胞比容远低于全身水平,且会随充血升高而增加,进而提高氧输送量(O2)”这一事实,在约半个世纪后才被人们认识到。最后,克罗格认为氧扩散可利用全部毛细血管表面积,而非仅利用与红细胞直接接触的表面积。
尽管存在这些后续发现,克罗格通过测量氧在组织中的扩散,证明了他是一位卓越的实验科学家。借助100年前的现有技术,他进行了细致且准确的测量,所得结果与现代先进技术的测量结果相差不大——这无疑是一项了不起的成就。
假设四:毛细血管运动机制
毛细血管募集概念是克罗格获得诺贝尔奖的核心依据(Gjedde,2010),该概念认为:骨骼肌毛细血管必须具备收缩能力,能完全阻断红细胞和血浆流——诺贝尔奖委员会将这种能力称为“毛细血管运动调节机制”,克罗格本人则称之为“毛细血管运动功能”。
克罗格1919年的研究结果并未明确所谓“毛细血管运动调节机制”的细胞来源,但他在第三篇论文中探讨了这一问题(Krogh,1919c,第470页):
“有充分证据表明,毛细血管并非仅在压力作用下被动扩张,而是能主动扩张和收缩。(鲁热,1879)证实了瞬膜以及某些胎儿和幼虫毛细血管中存在可收缩毛细血管[……],并描述了一种分支细胞——这些细胞分布在可收缩毛细血管的外侧,他认为这是收缩元件。”
背景
Roy & Graham Brown(1880,引自Krogh,1919c)在研究青蛙蹼、舌头和肠系膜循环时指出:
“提高血管内压无法使毛细血管直径产生可测量的增加,但无论压力高低,毛细血管直径都可能自发变化。因此,毛细血管的弹性极小,而必须具备主动收缩能力。”他们还提到:“……毛细血管的管径在不断变化。”
至少从19世纪60年代起,毛细血管的收缩能力就一直是研究和争论的焦点。Stricker在青蛙瞬膜中观察到毛细血管管腔直径的自发变化,这种变化与内皮增厚同步,但毛细血管外径无减小(Stricker,1865)。1903年,Steinach和Kahn报道了此类毛细血管的“真性”收缩(管腔和外径同时减小)(Steinach & Kahn,1903),正如克罗格(1919c)所引用的:
“……(他们)观察到(毛细血管)自发收缩和扩张,尽管血管内压维持在0。通过刺激背交感神经可诱发收缩,且……在取自哺乳动物的多种器官毛细血管中也观察到了自发收缩。”
与Stricker的研究(Stricker,1865;Krogh,1919c)无关,Rouget在包括青蛙玻璃体膜在内的多种组织中,发现了沿毛细血管外表面分布的细胞体(Rouget,1873、1879)。尽管无法对这些细胞进行染色,他仍观察到了细长的原生质细胞突起——这些突起以类似平滑肌纤维的方式环绕毛细血管管腔,并推测它们是毛细血管水平的收缩细胞(Rouget,1879)。1902年,Mayer采用亚甲蓝对多种组织中的鲁热细胞(Rouget细胞)进行染色,发现毛细血管前动脉和小动脉上的环形平滑肌纤维,逐渐过渡为沿毛细血管管腔呈分支状分布的鲁热细胞突起(Mayer,1902)。
1919-1922年间,在克罗格实验室工作的Bjovulf J. Vimtrup,研究了青蛙瞬膜、舌头、蹼以及蝌蚪和其他两栖类幼虫尾部毛细血管的收缩能力(Vimtrup,1922)。他发现毛细血管直径的变化与毛细血管前动脉血压无关,且毛细血管自发收缩总是从管腔的某个特定位置开始,然后以不同速度扩散至整个毛细血管长度。收缩的起始位置与鲁热细胞体的分布位置一致,他还观察到:随着收缩扩散,鲁热细胞体、细胞突起以及下方的内皮细胞均出现形态变化。据此,他得出结论:毛细血管直径由鲁热细胞独立调控。
Vimtrup的实验既包括切片标本观察,也包括在体观察。关键在于:在观察青蛙舌头肌肉中的鲁热细胞和毛细血管收缩时,Vimtrup并未观察到能阻断红细胞通过的闭塞现象,仅观察到毛细血管收缩至使血细胞变形、呈更长形状的程度(Vimtrup,1922)。
现代视角
1922年,克罗格在撰写关于毛细血管的著作时认为,鲁热细胞通过其环绕毛细血管的原生质突起收缩,从而缩小管腔(Krogh,1922;Krogh等人,1922)。在随后的十年里,研究人员在多种物种和器官中,均持续观察到毛细血管直径的自发变化和可诱导变化。但未观察到毛细血管自发闭塞的现象。
当时的争议焦点在于:毛细血管直径变化是由毛细血管内皮细胞自身的收缩特性引起,还是由毛细血管周围的鲁热细胞引起。Zweifach(1934)认为,内皮细胞在机械刺激下可能会增厚并向管腔内隆起,从而闭塞毛细血管。
在克罗格实验室,Bensley证实了Vimtrup的形态学和功能性发现,包括鲁热细胞在活体青蛙舌头和蹼毛细血管收缩中的起始作用(Bensley & Vimtrup,1928)。在青蛙瞬膜标本中,他们观察到毛细血管对电刺激的反应:管径存在变化,但极少出现毛细血管闭塞(Bensley & Vimtrup,1928)。重要的是,即使出现管径变化,其速度也过慢(闭塞需15-20秒,重新开放需约45秒),无法匹配代谢需求的变化速度(Sanders等人,1940)。如前文所述(图6A),这种极慢的动力学特性与骨骼肌运动充血的动力学特性,或大鼠快慢肌纤维中小动脉血管运动调控的动力学特性(Behnke & Delp,2010)均不相符。
20世纪30年代,正如Sanders及其同事(1940)所总结的,“毛细血管收缩能力”的观点日益受到质疑,该领域随后分裂为两派:一派支持毛细血管主动收缩(例如Kahn & Pollak,1931;Sandison,1931),另一派认为毛细血管是纯粹的被动血管,无主动收缩能力。
后来,Zweifach在观察到肠系膜毛细血管起始端存在狭窄后,提出了“毛细血管前括约肌”这一术语。尽管在骨骼肌中从未在功能或解剖层面发现过这种结构,但它仍作为所有组织共有的结构出现在全球的医学生理学教科书中。
毫无疑问,研究对象涵盖的组织和物种范围过广(两栖类幼虫、青蛙、兔子),且“毛细血管”和“周细胞”缺乏“明确界定”,这些因素共同导致了认知混乱(Zweifach,1939;Sanders等人,1940;Sanders & Florey,1940)。值得注意的是,Zimmerman对多种组织中的鲁热细胞进行了全面分类,并为其赋予了如今的名称——“周细胞”(pericyte),同时还描述了动脉与静脉之间的三种过渡形态(Zimmerman,1923)。他使用“毛细血管前周细胞”一词来指代“位于动脉(或小动脉?)末端、连接毛细血管系统的周细胞”(Zimmerman,1923)。
Fulton和Lutz采用微电极刺激法,证实了青蛙咽后膜中“毛细血管起始端括约肌样结构”内周细胞的神经调控作用,他们的研究基于Zimmerman和Vimtrup对该收缩单元的分类(Fulton & Lutz,1940);后来Rhodin将这种结构称为“毛细血管前括约肌”(Rhodin,1967)。
如今,周细胞仍被认为是视网膜(Puro,2007)和大脑(Hamilton等人,2010;Hall等人,2014;但参见Hill等人,2015的相反观点)中毛细血管血流的主要调控者。Tilton研究了心脏和骨骼肌周细胞的超微结构(Tilton等人,1979a),并报道了大鼠骨骼肌周细胞对血管活性药物产生形态学反应的证据(Tilton等人,1979b)。近年来,研究人员还探讨了心脏周细胞在灌注不足和缺血状态下的收缩潜力(Methner等人,2019;O'Farrell等人,2017)。然而,尽管经过了一个多世纪的研究与推测(Attrill等人,2019),周细胞在骨骼肌中“匹配氧输送与氧摄取(即$dot{Q}text{O}_2$与$dot{V}text{O}_2$)”中的明确作用,仍未得到证实。
假设五
肌肉摄氧量从静息到运动状态的增加(完全)是通过募集更多肌肉毛细血管,进而缩短毛细血管间扩散距离实现的。
现代视角
若骨骼肌在静息状态下,多数毛细血管中就存在红细胞通量(见表1右侧),那么已形成血流的毛细血管内红细胞通量和血细胞比容的增加,必然是灌注性和扩散性氧通量升高的原因(图4)。此外,从静息到运动状态,肌内氧分压会显著降低(而非克罗格所认为的升高)(图1右侧、图2B),这会导致肌红蛋白部分去饱和,从而缩小功能性氧载体耗竭区域,提高肌细胞内氧扩散效率(综述参见Honig等人,1997)。
对肌肉组织间隙氧分压的测量进一步印证了这些现代观点——组织间隙氧分压介于微血管氧分压与独立测得的肌细胞内氧分压之间($ce{P_{O2mv}}$、$ce{P_{O2is}}$、$ce{P_{O2im}}$;Richardson等人,1995、2006;Hirai等人,2018、2019)。在犬类腓肠肌-跖肌复合体中,运动训练(使毛细血管密度升高)和肢体固定(使毛细血管密度进一步升高)对毛细血管密度的调控,与肌肉摄氧量相互独立,这支持了“毛细血管内和肌细胞内的变化(而非毛细血管密度本身)决定肌肉摄氧量”的观点(Hepple等人,2000)。
假设六:毛细血管的功能均一性
克罗格认为肌肉毛细血管具有二元功能:要么收缩并阻断血浆和红细胞流(无氧气输送),要么开放并维持固定的氧气输送量——且不同毛细血管的氧气输送量无差异,静息与收缩状态下的氧气输送量也无变化。
现代视角
在哺乳动物骨骼肌中,毛细血管直径范围为2~8微米,长度范围为50~500微米甚至更长(Mathieu-Costello等人,1989;Poole等人,1989、1997;Sarelius,1986)。如今已明确,无论是静息还是收缩状态,毛细血管的红细胞通量、流速、血细胞比容,以及红细胞路径长度和通过时间均存在极大异质性(Sarelius,1986、1990;Sarelius & Duling,1982;Damon & Duling,1984、1985),且毛细血管红细胞分布和血流在一定程度上依赖于肌肉长度(Kindig & Poole,1999、2001)。
即便在静息和收缩状态下,由同一终末小动脉供血的相邻毛细血管之间,这种异质性也依然存在。此外,快肌和慢肌纤维的毛细血管血流模式差异显著,例如胫骨前肌的红细胞流速高于比目鱼肌(Dawson等人,1987)。而且,肌肉收缩开始后,红细胞通量、流速和血细胞比容都会迅速(约1~2秒)显著增加(Kindig等人,2002)。
这一观察结果至关重要:从克罗格的二元视角来看,静息时红细胞(和血浆)通量极低的毛细血管虽未被“募集”,但在运动过程中,这些毛细血管会为氧气输送量和肌肉摄氧量的提升发挥作用(图2A、图10A;Kindig等人,2002;Poole等人,2011、2013;Poole,2019)。

图10 肌肉中血液-肌细胞间氧气或底物交换的增加,无需通过新的毛细血管募集来解释
A图:简化模型中,毛细血管底物输送量与肌肉底物摄取量的关系接近指数曲线(左图)。克拉克及其同事采用1-甲基黄嘌呤代谢法(例如Rattigan等人,1997a、b;Youd等人,1999),基于该模型计算收缩骨骼肌的毛细血管募集情况。该模型的核心假设为:(1)微循环血流(毛细血管层面)呈均一分布;(2)静息状态下,所有毛细血管均处于曲线的平台区。
在这些研究中,从静息到收缩状态下1-甲基黄嘌呤代谢量的任何增加,都被视为毛细血管表面积扩大的标志,而毛细血管表面积扩大又被认为是由原本无血流的毛细血管被募集所致。然而,如正文所述,实际情况是:无论静息还是收缩状态,微血管血流均存在显著异质性。
因此,右图将毛细血管分为两组:低血流组和高血流组——这一分类恰好反映了毛细血管床中实际存在的部分异质性(参见正文及支持视频S1:该视频显示健康状态下,绝大多数肌肉毛细血管存在红细胞通量,而心力衰竭状态下则不然)。在这个简化模拟中,低血流组毛细血管处于底物摄取的血流限制区,高血流组毛细血管处于曲线的平台区(“扩散/表面积限制区”);但需说明的是,具体表现会因所研究的底物种类以及肌肉代谢活动设定的边界条件而异。
静息状态下的平均血流接近曲线平台区(左图白色箭头,与克拉克等人的模型一致)。两组毛细血管的血流均增加后——收缩状态下的平均毛细血管血流用右图黑色箭头表示(与克拉克等人模型中的血流相当),此时氧气或底物摄取量(提取量)的升高,与“毛细血管募集”模型的预测结果完全一致。这表明:在无任何毛细血管募集的情况下,异质性血流分布的毛细血管中血流增加,是对克拉克等人实验数据更合理、更符合生理实际的解释。直方图总结了这一结论。此外,该模型(无需毛细血管募集)与从静息到运动状态下通过活体显微镜观察到的数据相符(例如Kindig等人,2002)。
B图:毛细血管血流均一化、血细胞比容升高以及纵向募集(图12核心特征),能使血液-肌肉氧气扩散可用的表观毛细血管表面积显著增加,其效果优于克罗格所假设的“原本关闭的毛细血管开放”(Jespersen & Østergaard,2012;Angleys & Østergaard,2020)。绿色和蓝色曲线分别代表毛细血管血流均一化和血细胞比容升高处于两种中间程度时,血流与肌肉摄氧量($ce{dot{V}_{O2}}$)的关系(相对于静息状态)。需注意:从生物物理学角度看,这些机制可通过血流成比例增加,满足肌肉摄氧量升高的需求。
相反,该图也解释了为何不能将组织中可扩散示踪剂的稳态浓度,作为“原本关闭的毛细血管开放”的证据。红色箭头表示:在无上游血流限制的情况下,某些因素(如糖尿病)导致的毛细血管形态和功能改变,可能通过干扰毛细血管血流模式和/或红细胞纵向募集,限制血液-肌肉氧气运输。这一特征体现了两种微循环模型在概念和功能上的重大差异:一种是“毛细血管均开放但灌注量不同”(参见图12),另一种是“部分毛细血管关闭”(克罗格模型)。
如近期综述所述(Poole,2019),某一毛细血管支持血液-肌细胞或肌细胞-血液间氧气(及底物)运输的能力,在很大程度上取决于以下因素:毛细血管容积和功能性表面积、红细胞通量、流速和血细胞比容,以及红细胞路径长度和通过时间。
此外,交换分子的具体特性也很重要:例如,是由红细胞携带(如氧气)还是由血浆携带(如葡萄糖、游离脂肪酸);血液-组织间隙-肌细胞间的压力差或浓度差大小;是否涉及特殊转运过程(如葡萄糖转运,Wasserman等人,2011;McClatchey等人,2019)。
重要的是,正如图10(上图)的理论分析结论所示,Bentzer等人(2001)已证实:骨骼肌中毛细血管滤过系数(即膜通透性与表面积的乘积),与被灌注毛细血管的数量相互独立。
基于上述考量,“毛细血管是氧气或底物输送的固定单位”这一概念显然是错误的。图10A表明:无需任何新的毛细血管募集,仅通过增加已形成血流的毛细血管中的血流和交换量,即可实现完全相同的血液-肌肉氧气(或其他底物)扩散通量。这一计算本身就证明:无论健康还是疾病状态下,要解释血液-肌细胞扩散通量的变化,克罗格的毛细血管募集模型并非必需(图10B;另参见Angleys & Østergaard,2020)。
假设七
“横纹肌横截面上每平方毫米的毛细血管数量,似乎是代谢强度的函数——小型哺乳动物的毛细血管密度高于大型哺乳动物。”(Krogh,1919a)
“……显然,在这些情况下,参与循环的血管数量远超氧气供应所需。因此,血管开放数量的增加,很可能是为了满足肌肉的其他需求。”
现代视角
这一观察结果后来被进一步拓展:不仅限于物种间差异,慢性电刺激、运动训练导致的肌肉代谢能力动态提升,以及心力衰竭、糖尿病等疾病引发的功能障碍,均会影响毛细血管密度(综述参见Hudlicka等人,1982、1987;Saltin & Gollnick,1983;Hudlická,1985、2011;Haas & Nwadozi,2015;Parise等人,2019;Poole等人,2012;Hirai等人,2015)。
人们还认识到:肌肉毛细血管床的功能不仅是输送氧气和底物;对于快肌 glycolytic(糖酵解型)纤维而言,它还需有效清除代谢产物,并可能参与肌因子的转运。有趣的是,快肌纤维对氧气输送量与氧气摄取量($ce{dot{Q}_{O2}/dot{V}_{O2}}$)比值的调控,使得其微血管和组织间隙氧分压低于慢肌纤维(图6B;Dawson等人,1987;Behnke等人,2003;McDonough等人,2005;Hudlická,2011;Poole等人,2020a)。
克罗格的其他重要观察(1919a、b、c)
(1)毛细血管结构
“静息状态下被拉伸至正常长度的肌肉中,毛细血管大致呈直线状;而收缩状态下的肌肉中,毛细血管会变得高度弯曲;毛细血管之间存在一定程度的吻合,围绕肌纤维形成长网状结构。”(Krogh,1919a,第410页)
现代视角
约50年后,Gray及其同事(1967)发现犬类肌肉等长收缩时,毛细血管会发生弯曲和盘绕;Aquin & Banchero(1981)观察到犬类腓肠肌中的毛细血管呈螺旋状环绕肌纤维,并指出这种几何结构有利于血液-肌细胞间的扩散。
与大脑中毛细血管呈各向同性(无偏好取向)不同,骨骼肌毛细血管呈各向异性,且与肌纤维方向高度一致。但部分肌肉的毛细血管可能存在明显的弯曲和分支,有时类似多分支结构——例如家鸽胸大肌(Mathieu-Costello,1991)和金枪鱼的高有氧能力肌肉(图11)。
Mathieu-Costello将毛细血管弯曲度定义为肌节长度的函数(Mathieu-Costello等人,1989、1991、1994;Poole等人,1989;Poole & Mathieu-Costello,1992)。考虑到毛细血管床的弯曲度和分支特征(图11),血液-肌细胞氧气扩散的更适宜模型是希尔圆柱体模型(Hill cylinder),而非点源模型(克罗格-厄兰模型,公式(1)(2),图1左侧、中间)。
希尔模型(Ellis等人,1983)所描述的氧气输送系统效率更高:由于肌纤维表面与毛细血管接触的比例更大,随着与某一毛细血管距离的增加,依赖该毛细血管供氧的肌纤维体积会减少。这与克罗格模型(克罗格-厄兰模型)形成鲜明对比——在克罗格模型中,与毛细血管距离越远,组织体积越大(参见上文公式(1))。
此外,对大鼠、蝙蝠和蜂鸟的肌纤维与毛细血管几何结构的跨物种比较表明:对于高有氧能力肌肉的氧气扩散能力而言,重要的是肌纤维周围毛细血管的数量和容积,而非扩散距离本身。当然,这些毛细血管中红细胞的数量也至关重要。
有趣的是,Glancy及其同事(2014)发现毛细血管可能实际嵌入肌纤维肌膜中,这或许能使氧气和底物优先输送到特定肌纤维。
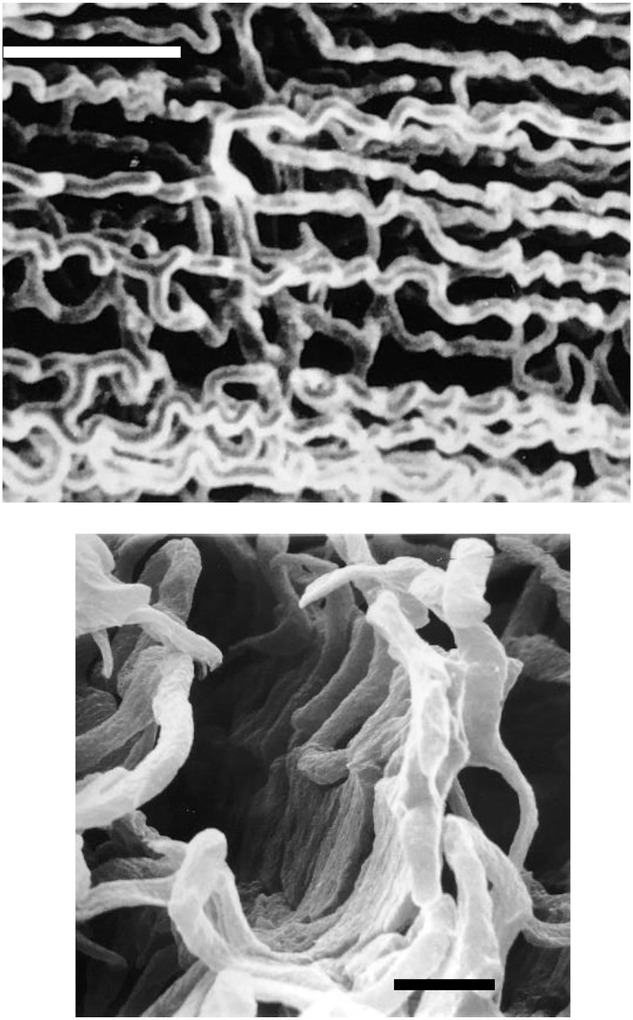
图11 毛细血管的弯曲和分支有利于血液-肌细胞间氧气和底物的扩散
腐蚀铸型显微照片展示了毛细血管结构的极端案例——这些结构增加了毛细血管与肌纤维的表面接触面积,从而促进血液-肌细胞间氧气和底物的扩散,并支持希尔氧气扩散模型(而非克罗格模型);肌肉缩短时毛细血管弯曲度增加,也符合这一特征(Ellis等人,1983、1990):
上图:高度弯曲且分支的骨骼肌毛细血管床(家鸽胸大肌;比例尺=50微米);
下图:金枪鱼红色肌肉(比例尺=10微米)。
毛细血管弯曲度是肌节长度的函数。在某些物种的高氧化型肌肉中,毛细血管的分支程度可能更为显著。图源自Poole(2019)。
(2)毛细血管-小动脉信号传导
克罗格提出,毛细血管具有内在信号机制,可引起上游小动脉扩张,从而增加自身灌注量。这一极具洞察力的观察,启发了后续几代生理学家运用先进技术,探索该反应的特征和作用。
杜林及其同事在一篇综述中指出:“……功能性充血是一个复杂过程,涉及毛细血管、小动脉和小静脉”(Duling等人,1987)。研究发现,河豚毒素(10⁻⁶mol/L)无法阻断相关血管扩张,因此推测该过程涉及平滑肌细胞和/或内皮细胞的直接细胞间偶联及缝隙连接,而非神经调控(Segal & Duling,1987、1989)。
随后,McGahren等人(1998)发现证据表明:毛细血管内皮细胞膜电位(乙酰胆碱可使其超极化,去氧肾上腺素可使其去极化)会对组织间隙环境的变化做出响应。如今已明确,微血管内皮细胞与平滑肌细胞之间存在电偶联(Xia等人,1995),且信号可通过内皮细胞沿微血管传导,激活电压依赖性钠通道和钙通道(α1H亚型,CaV3.2),进而引发小动脉扩张(Figueroa等人,2007)。
这些结果(另参见Beach等人,1998;Bagher & Segal,2011)为克罗格数十年前提出的“毛细血管与小动脉间存在功能性电信号通讯”提供了在体证据。
(3)毛细血管形态
关于不同组织类型间毛细血管形态功能的差异,克罗格指出:“有必要强调,为不同器官供血的血管,其功能在数量上甚至质量上都存在差异”(Krogh,1921)。与同时代研究者一样,克罗格的结论广泛适用于不同物种和器官,但他也认识到毛细血管存在显著的异质性。
不同器官对其毛细血管的结构有截然不同的要求。例如,肠系膜毛细血管(Zweifach,1939)、蝙蝠翼膜毛细血管(Salathe,1977)和肺部毛细血管(Pande & Hughes,1983)——这些都经过了深入研究——均缺乏骨骼肌毛细血管所具备的、与相邻肌纤维间的结构支撑(Caulfield等人,1979;Borg & Caulfield,1980)。因此,这些毛细血管对跨壁压力更为敏感:当血管外腔或血管腔内出现极小的正压时,其单薄的单细胞管壁会分别发生塌陷或扩张。
同理,我们不应认为“用于跨毛细血管溶质吸收的有孔内脏和肠道毛细血管”与“通过散热调节核心体温的皮肤毛细血管”具备相同的调控特征。这种差异也适用于骨骼肌:其对氧气、底物输送及代谢产物清除的需求,会在两个数量级范围内快速动态变化(综述参见Poole,2019;Poole & Jones,2012)。或许正是由于广泛研究了不同物种、不同器官的毛细血管,克罗格有时非常清楚:将不同毛细血管床的结构和功能原理统一化存在风险(Krogh,1922)。
从克罗格绘制的图9(Krogh,1920b,1922)中可明确看出:该图源自青蛙肌肉,显示的血管直径异常粗大,是其终末小动脉直径的数倍——这显然并非哺乳动物肌肉中常见的结构。
毛细血管募集假说:科学哲学视角
在不受干扰的情况下,科学本质上具有自我修正的特性(Whipp,2010),正如波普尔(Popper,1934)所言:“对‘必须正确’的渴望,暴露了对科学的错误认知”。但如果有强大的影响力决定了权威机构将接受哪些实验结果或数据,情况会如何?例如,13世纪教会拒绝接受某些发现,仅仅因为这些发现“未在亚里士多德的著作中出现”(如Principe,1985;Whipp,2010)。如果不存在明确的“反派”,反而是“最崇高的意图”阻碍了科学发现,又该如何?
丹尼尔·J·布尔斯廷(Daniel J. Boorstin)在其巨著《发现者》(The Discoverers)中调侃道:“科学进步的最大障碍不是无知,而是自以为知的幻觉”(Boorstin,1983)。试想,当世界最高科学荣誉——诺贝尔奖——被视为对某项发现、概念或理论的绝对认可时,这可能会阻碍该领域后续的发展——这显然与奖项设立的初衷背道而驰。
如上所述,克罗格因发现“毛细血管运动系统”(即毛细血管自身的主动收缩功能)而获得诺贝尔奖,他认为该系统调控着骨骼肌中的红细胞通量,进而调控氧气输送。若上述“奖项阻碍发展”的情况属实,对科学而言是悲剧,对克罗格而言则是双重悲剧——因为他最核心的科学原则就是对观察准确性、解读与报告诚实性的极致追求。
事实上,人们不禁会思考:若玻尔(Bohr)曾因“发现肺内主动氧气分泌”而获诺贝尔奖,克罗格(及其妻子玛丽)推翻该假说的“七个小恶魔”系列论文(Krogh,1910a-e;Krogh & Krogh,1910a,b)还能获得如此广泛的认可吗?要知道,克里斯蒂安·玻尔(Christian Bohr)曾三次获得诺贝尔奖提名。
如上所述,与克罗格证明“无论静息、运动还是高原环境下,肺内血气屏障的氧气主动转运均非必需”类似,收缩骨骼肌中血液-肌细胞氧气扩散通量的增加,也不需要通过“新募集(即开放)原本无血流的毛细血管”来实现。
克罗格的卓越洞察力为新的思考和研究方向提供了启发,但本文重点讨论的他那几篇“诺奖论文”(Krogh,1919a-c),或许并未如诺贝尔奖委员会所期望的那样,成为科学发现的跳板。例如,时至今日,仍有研究者教条地坚持克罗格的毛细血管募集理论,且往往从未观察过骨骼肌的毛细血管床(完全违背“不盲从权威”的原则),这阻碍了我们对“健康与疾病状态下微血管调控及血液-肌细胞氧气、底物扩散”机制的理解(Poole,2019)。
毛细血管募集假说:病理生理学视角
血管疾病约占全球死亡人数的三分之一,且会导致严重残疾。目前“血管疾病”的定义侧重于影响动脉和小动脉形态与功能的疾病——这些疾病会限制血液供应,进而减少下游细胞的含氧血液供应。
克罗格研究最重大的遗产或许是他最早认识到:运动时肌肉中毛细血管的血液分布必须发生改变,才能满足细胞氧气摄取量的同步增加。尽管克罗格提出的调控机制被证明不成立,但他的初始观察揭示了整个毛细血管床在组织氧气供应及疾病进程中的作用。
具体而言,若某些因素(如毛细血管血流均一化、纵向募集、血细胞比容升高、糖萼功能等)无法实现“毛细血管内红细胞数量增加”,其对组织氧气净转运的限制作用,可能与“血液供应大幅减少”相当。
目前的诊断方法可让医生观察大多数器官的大血管和局部血流灌注,从而了解衰老、血管危险因素及疾病对阻力血管的影响。然而,这些方法的分辨率无法捕捉毛细血管的血液分布和血流情况,因此我们无法确定:衰老、血管危险因素及疾病对毛细血管形态和功能的已知影响,是否会在“单纯影响血液供应”之外,进一步限制细胞获取氧气。
事实上,在患有脑血管疾病且因大血管狭窄导致局部氧气供应减少的患者中,只有考虑微血管血流紊乱的影响,才能解释脑氧气摄取量的变化。与此同时,阿尔茨海默病(AD)患者不存在大血管病变,这使得大多数研究者和医生排除了“血管疾病变化在其发病机制中起作用”的可能性——尽管阿尔茨海默病与心血管疾病和中风共享大多数危险因素。
然而,近期研究发现阿尔茨海默病患者存在毛细血管血流紊乱,这表明脑组织中氧气水平不足。这一发现说明,我们对组织氧气转运的理解不完整,误导了我们对疾病基本机制的研究方向。
因此,克罗格通过生物物理模型和细致测量来解释肺和肌肉氧气转运的努力仍在继续,如今的目标是探索:我们目前对“血管疾病”的认知,是否仅仅是尚未探索的“冰山一角”。
结论
在研究肌肉毛细血管之前,克罗格夫妇(奥古斯特与玛丽)最卓越、影响最深远的研究是1910年发表在《斯堪的纳维亚生理学档案》(Skandinavishe Archiv. Fur Physiologie)第23卷上的“七个小恶魔”系列论文(Krogh,1910a-e;Krogh & Krogh,1910a,b)。
正如福斯特(Forster,1996)所述,这些“小恶魔”通过克罗格改进的气体张力计/微量张力计证明:动脉血氧分压低于肺泡血氧分压,因此肺内氧气的主动转运或分泌并非必需。这一结论与他的导师克里斯蒂安·玻尔(Christian Bohr)以及著名英国呼吸生理学家约翰·斯科特·霍尔丹(John Scott Haldane)的观点相悖——后者直到1936年仍坚信肺内存在主动氧气分泌(Haldane,1936)。
然而,“主动过程不参与肺氧气摄取”这一结论,也影响了克罗格提出的毛细血管募集理论——该理论旨在解释收缩肌肉中血液-肌细胞氧气扩散通量的增加。
他们在研究肺部时面临的问题之一是:静息状态下测得的肺氧气摄取量,不足以满足高强度运动或高原环境下的巨大需求。1915年,玛丽·克罗格(Marie Krogh)证明运动时肺氧气摄取量会显著增加;如今我们知道,这一过程通过多种机制实现,包括血管募集与扩张、肺毛细血管血容量增加——这些机制由心输出量和肺血管压力升高驱动(综述参见Cerretelli & di Prampero,1997)。
换言之,尽管没有证据表明存在跨毛细血管氧气分泌,但其他主动生理过程会导致氧气摄取量增加——其中部分(而非全部)依赖于毛细血管募集(发生在肺部,而非骨骼肌)。正如杰德(Gjedde,2010)所认为的,克罗格和玻尔关于肺氧气摄取的观点都有部分正确性,但生理学家花了数十年时间才广泛接受这一观点。
1920年,克罗格的诺奖论文(Krogh,1919a-c)将“毛细血管运动”或“毛细血管募集”理论与厄兰(Erlang)的组织氧气摄取数学模型相结合,彻底改变了人们对毛细血管的认知。毛细血管不再被视为动脉和小动脉下游的被动“仆人”,而是能根据相邻肌细胞的代谢需求主动收缩和舒张。
如上所述,该模型基于一系列假设(见下文),部分源于以下技术局限:墨水灌注法标记开放毛细血管存在困难,以及在体实验中使用深度乌拉坦麻醉。这些限制可能使克罗格的实验更易观察到“毛细血管募集”现象。值得注意的是,其实验室后续由维姆特鲁普(Vimtrup)、本斯利(Bensley)等人开展的研究,并未观察到毛细血管关闭的现象。
以下这些假设,如今已在活肌研究中被确凿推翻(斜体部分):
1. 静息状态下,肌细胞内部分区域的氧气分压接近零或为零。实际上,静息时肌细胞内氧气分压远高于0 mmHg,且运动时会降低(而非升高)。
2. 毛细血管总容积超过血液总容积。更符合实际的估计是,肌肉毛细血管总容积≤血液总容积的10%。
3. 氧气分压随与毛细血管距离的增加而系统性降低。氧气分压的最大降幅出现在从毛细血管到肌膜的过程中;肌细胞内氧气分压分布均匀,无明显梯度。
4. 毛细血管具有收缩能力,可主动收缩和舒张。目前没有令人信服的证据表明骨骼肌毛细血管具备这一特性。
5. 在某一特定肌肉中,每条毛细血管供应的组织容积相同,且随与毛细血管距离的增加而增大。考虑到毛细血管及网络的几何结构,氧气输送的最佳模型是希尔圆柱体模型——该模型中,组织容积随与毛细血管距离的增加而减小,是一种效率高得多的系统。
6. 每条毛细血管在静息和运动状态下的氧气输送能力相同(即特定毛细血管内的红细胞通量和流速不会增加)。无论静息还是运动状态,不同毛细血管间的红细胞通量、流速和血细胞比容均存在显著异质性(运动时这三项指标均会升高)。将每条毛细血管视为固定的氧气输送“量子单位”,既无科学依据,也无实验证据支持。
7. 从静息到运动,肌内氧气分压保持不变(即未认识到肌红蛋白在氧气转运中的作用,也未认识到毛细血管血细胞比容会升高)。从静息到运动,肌肉氧气摄取量会显著增加,这源于毛细血管血细胞比容升高、毛细血管交换表面积纵向募集以及肌红蛋白去饱和。
8. 肌肉氧气摄取量的动态范围约为10倍。利用丹麦克罗格研究所开发的安德森-萨尔廷(Andersen & Saltin,1985)恒输注热稀释技术,理查森等人(Richardson,1993)证明:人类肌肉氧气摄取量的动态范围至少为100倍。
人们不禁会思考:若克罗格能获得本文所述的数据(尤其是图1-3、图6,以及图12中当代肌肉毛细血管功能模型),结果会如何?他很可能会像我们一样,对美丽的、呈链状互连的线粒体网(图2B右插图;Bakeeva等人,1978;Glancy等人,2015;Vincent等人,2019)感到惊叹——这一结构可能会推翻“肌细胞内扩散距离”的整个概念。
同样,用希尔氧气转运模型(Ellis等人,1983;图11)替代克罗格-厄兰模型(Krogh,1919a,c),也会带来类似的认知转变。新的技术表明:运动时,犬类和人类肌肉中从微血管到组织间隙的氧气分压降幅显著,且收缩肌细胞内的氧气分压极低(且可能均匀分布)——这些发现均支持上述新认知(图1、图2、图7;Honig等人,1997;Richardson等人,1995,2006)。
最后,尽管数十年间研究不断,但仍缺乏静息状态下毛细血管收缩的有力证据;同时,大量研究表明人类肌肉运动时,毛细血管血红蛋白浓度仅轻度升高(这一观察结果排除了“显著毛细血管募集”的可能性,如Davis & Barstow,2013;Okushima等人,2015,2016)——这些事实肯定会促使克罗格质疑并修正他的毛细血管募集理论。
事实上,他的前学生兼同事汉斯·乌辛(Hans Ussing)教授(克罗格曾教他吹制以其名字命名的“乌辛室”)曾说:“如果克罗格也会犯错,那我们也可能犯错……”(Lindsten,2001)。这句话体现了克罗格所认同的灵活性——他认为,当实验结果超出预期时,质疑或修正假设至关重要。
图12 毛细血管功能的当代模型:静息与运动状态
该图总结了“无需募集原本无血流的毛细血管,仅通过已存在红细胞流的毛细血管,即可实现从静息到运动状态下血液-肌细胞氧气(及底物)扩散通量增加”的机制:
A. 原本低血流毛细血管中的血流(红细胞通量)显著升高,意味着这些在静息时对血液-肌细胞扩散无足轻重的毛细血管,在运动时变得重要。
B. 微血管在三维空间中紧密相邻,使得血管间可发生氧气扩散(Pittman,1995,2011;Popel,1980,1982)——理论上,这有助于缓解氧气输送与摄取不匹配的问题(插图由Mal Rooks Hoover,CMI提供)。
C. 毛细血管内红细胞数量(血细胞比容)升高,会增加灌注性和扩散性氧气传导率。需注意,这一效应在数量上可解释从静息到运动状态下血红蛋白+肌红蛋白总浓度信号的全部增加(Lutjemeier等人,2008)。
D. 红细胞流速增加且氧气提取分数提高,意味着毛细血管长度方向上更多的交换表面积会参与血液-肌细胞扩散(即“毛细血管纵向募集”)。
E. 血管内皮细胞表面层(被认为是“毛细血管血细胞比容低于全身水平”的关键因素)在高血流条件下可能发生改变。这一效应对血液-肌细胞氧气及底物扩散的影响尚待研究。
F. 从静息到运动,细胞内氧气分压急剧下降,这至少能维持(甚至可能增加)血液-组织间隙-肌细胞间的氧气分压差;同时,肌红蛋白去饱和,消除了“功能性氧气载体耗竭区域”(Honig等人,1997),提升了其氧气转运能力,进而提高细胞内氧气扩散能力(比例尺=1微米,Hirai等人,2018)。
克罗格会如何看待那些“坚持用毛细血管募集理论解释运动或高胰岛素状态下血液-肌细胞扩散通量增加,并继续绘制肌内氧气分压理论分布图”的研究者?克罗格本人肯定会全面权衡反对其假说的证据,并坚持在现代实验条件下对毛细血管床进行在体观察——遵循英国皇家学会的格言“不盲从权威”(Nullius in Verba),即“不轻易相信他人的话,亲自验证”,这些研究者也应如此。
根据我们目前的认知,通过超声造影(CEU)、1-甲基黄嘌呤转化曲线等技术对微循环进行的间接研究,并未发现毛细血管募集的证据。这些技术对“毛细血管募集”的解读,已不再得到现有最佳数据的支持。
倘若克罗格那几篇里程碑式的论文(Krogh,1919a-c)能真正如其预期的那样,成为科学发现的跳板,如今我们对肌肉微循环功能的理解会达到何种程度?可以肯定的是,我们对“微循环功能障碍在心力衰竭、糖尿病等疾病中的作用”以及“相关治疗对策”的研究,或许会取得更大进展。
克罗格的深远遗产
如今,克罗格众多的科学贡献仍频繁出现在丹麦报纸及全球各地的科学领域讨论中。他的名字是“测量与观察准确性”“实验设计精巧性”以及“对人类和多种物种各类生理过程及重要问题的非凡洞察力”的代名词(参见同期评论文章;Poole等人,2020b)。1970年,奥古斯特·克罗格研究所(August Krogh Institute)在哥本哈根成立,他的前学生们继续推进他开创的研究工作。每届国际生理科学大会(International Congress of Physiological Sciences)都会设立“奥古斯特·克罗格讲座”(August Krogh Lecture);1994年,他的女儿博迪尔(Bodil)主讲了美国生理学会(American Physiological Society)首届“奥古斯特·克罗格杰出讲座”(August Krogh Distinguished Lecture)。大约在同一时期,美国生理学会主席布莱恩·R·杜林(Brian R. Duling)教授向博迪尔透露,他实验室的门牌上,奥古斯特·克罗格的名字仍被列为在研研究员!
参考文献
Poole DC, Pittman RN, Musch TI, Østergaard L. August Krogh's theory of muscle microvascular control and oxygen delivery: a paradigm shift based on new data. J Physiol. 2020 Oct;598(20):4473-4507.
本文原作者简介
戴维·C·普尔(David C. Poole):堪萨斯州立大学(Kansas State University)运动机能学系及解剖学与生理学系教授,“大学杰出教授”(University Distinguished Professor),科夫曼杰出大学教学学者讲座教授(Coffman Chair for Distinguished University Teaching Scholars),拥有利物浦约翰摩尔斯大学(Liverpool John Moores University)理学博士学位(Scientiae Doctor)。
罗兰·N·皮特曼(Roland N. Pittman):弗吉尼亚联邦大学(Virginia Commonwealth University)医学院生理学与生物物理学教授、研究生项目主任,任职于弗吉尼亚州里士满市。
蒂莫西·I·马斯奇(Timothy I. Musch):堪萨斯州立大学运动机能学系及解剖学与生理学系“大学杰出教授”,与戴维·普尔共同主持该校克拉伦堡心肺实验室(Clarenburg Cardiorespiratory Laboratory)。
莱夫·厄斯特高(Leif Østergaard):奥胡斯大学(Aarhus University)伦德贝克基金会教授(Lundbeck Foundation Professor)、功能整合神经科学中心(Center of Functionally Integrative Neuroscience)主任,同时担任丹麦奥胡斯大学医院(Aarhus University Hospital)神经放射学研究室顾问及主任,丹麦皇家科学与文学院(Danish Royal Academy of Sciences and Letters)院士。
上述作者共同的研究方向包括:微循环功能、氧气与底物转运及细胞能量代谢,尤其关注静息与运动状态下肌肉的相关机制,以及健康与疾病状态下大脑的相关机制。
转载本文请联系原作者获取授权,同时请注明本文来自孙学军科学网博客。
链接地址:https://wap.sciencenet.cn/blog-41174-1503473.html?mobile=1
收藏