博文
锂缺乏与阿尔茨海默病的发病  精选
精选
|
锂缺乏与阿尔茨海默病的发病
阿尔茨海默病(AD)早期的分子变化尚不明确¹、²、³、⁴、⁵。本文研究表明,内源性锂(Li)在大脑中受到动态调节,并有助于在衰老过程中维持认知功能。在我们分析的金属中,锂是唯一在轻度认知障碍(MCI,AD的前期状态)患者大脑中显著减少的金属。在AD患者中,淀粉样蛋白的封存进一步降低了锂的生物利用度。我们通过在野生型和AD小鼠模型的饮食中去除锂,探索了内源性锂在大脑中的作用。将内源性皮质锂减少约50%,显著增加了β-淀粉样蛋白的沉积和磷酸化tau蛋白的积累,并导致促炎性小胶质细胞激活、突触、轴突和髓鞘丢失,以及认知功能加速衰退。这些效应至少部分是通过激活激酶GSK3β介导的。单核RNA测序显示,锂缺乏导致多种脑细胞类型的转录组变化,这些变化与AD中的转录组变化重叠。乳清酸锂(一种与淀粉样蛋白结合力较低的锂盐)替代疗法可预防AD小鼠模型和衰老野生型小鼠的病理变化和记忆丧失。这些发现揭示了内源性锂在大脑中的生理作用,并表明锂稳态的破坏可能是AD发病机制中的早期事件。使用可避开淀粉样蛋白的锂盐进行锂替代,是预防和治疗AD的一种潜在方法。
Aron L, Ngian ZK, Qiu C, Choi J, Liang M, Drake DM, Hamplova SE, Lacey EK, Roche P, Yuan M, Hazaveh SS, Lee EA, Bennett DA, Yankner BA. Lithium deficiency and the onset of Alzheimer's disease. Nature. 2025 Aug 6.
要确定AD的可治疗病因,需要从根本上理解导致记忆丧失的致病过程。尽管在确定与AD风险相关的基因变异方面取得了重大进展,但影响疾病发病时间的环境因素尚不明确¹、⁶。已发现一些与饮食、生活方式和环境相关的因素,但其对AD发病机制的作用尚不清楚¹、⁶、⁷。金属稳态的改变就是这样一个因素⁷、⁸、⁹、¹⁰、¹¹、¹²。这些研究主要集中在铁、铜和锌等金属的毒性作用上,在模型系统中,这些金属可促进β-淀粉样蛋白(Aβ)聚集、tau蛋白磷酸化或氧化应激⁶、⁷、⁸、⁹、¹⁰、¹¹、¹²。然而,金属在大脑功能中也具有重要作用,而AD中这种正常生理功能的破坏相对未被探索。
轻度认知障碍和阿尔茨海默病中的锂缺乏
为了探索金属离子稳态在AD中的作用,我们使用电感耦合等离子体质谱法(ICP-MS)评估了无认知障碍(NCI)的老年人以及遗忘型MCI或AD患者的大脑和血液中的27种常量和微量金属。测定了在AD中显著受累的前额叶皮层(PFC)和相对未受累的小脑中的金属水平。在所有被调查的金属中,只有锂在MCI和AD患者的PFC中显示出显著降低的水平(图1a、b和补充表1)。MCI和AD患者PFC中锂的皮质与血清比率的平均值和中位数,以及总皮质锂均显著降低(图1c、d),而小脑则无此现象(扩展数据图1a、b)。在第二个独立队列中,AD患者PFC中的锂水平也显著降低(图1e)。相比之下,MCI和AD患者的平均血清锂水平与对照组无显著差异(扩展数据图1c)。在本研究中,锂水平未受性别或死后间隔时间范围的显著影响(见方法)。其他几种金属的皮质与血清比率在AD中也发生了变化,但在MCI中没有(图1a、b和补充表1)。然而,锂的变化在所有被分析的金属中调整后的P值最低(图1b)。总之,这些结果表明,MCI和AD患者大脑中的内源性锂稳态受到干扰。
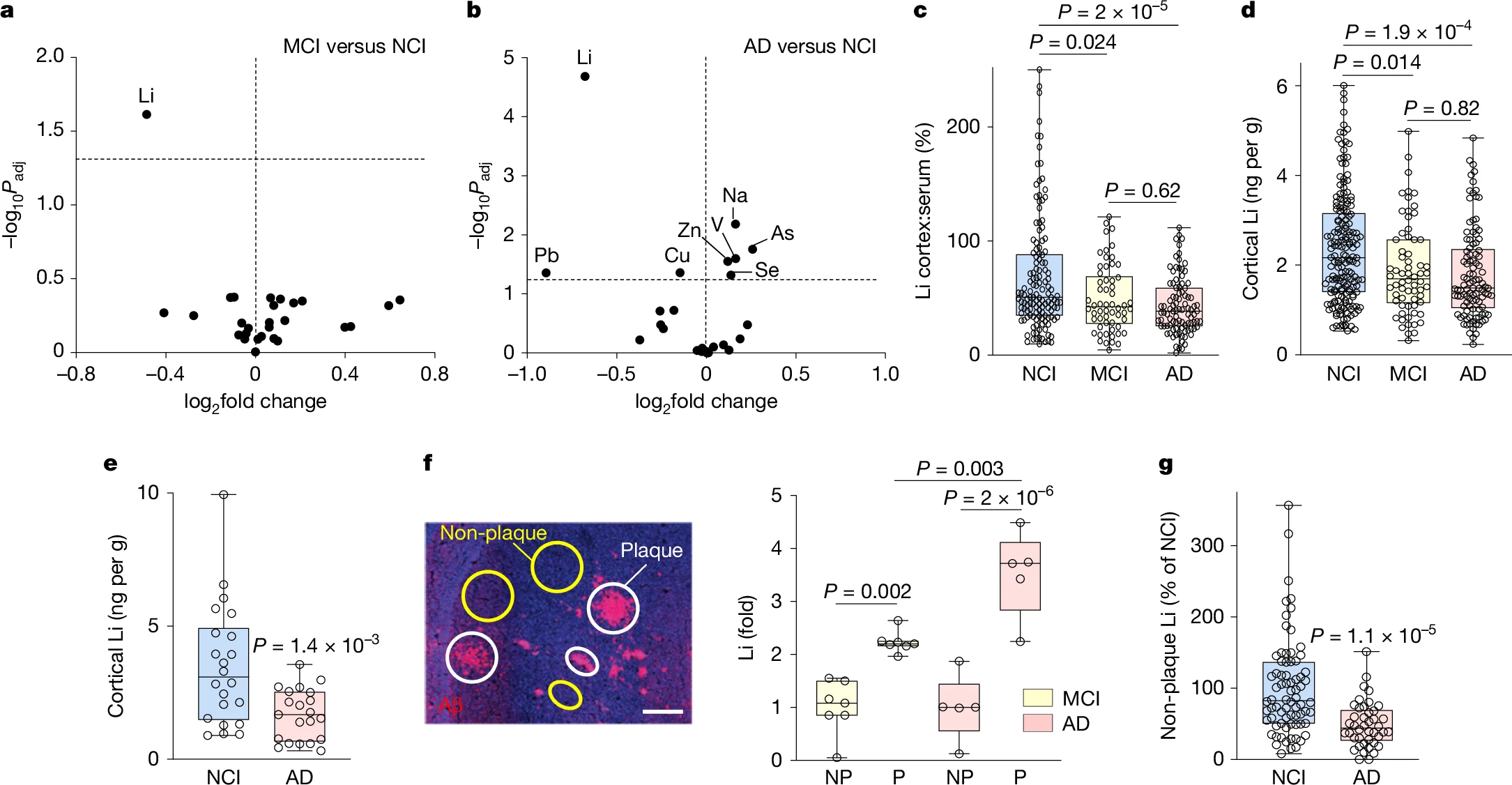
图1:锂缺乏与AD的发病
a、b,火山图显示MCI与NCI(a)以及AD与NCI(b)病例的PFC中金属皮质与血清比率的变化,及其统计显著性,通过单因素方差分析(ANOVA)与Tukey事后检验确定,随后对所评估的金属数量进行Benjamini-Hochberg校正。c、d,ROSMAP病例中的锂皮质与血清比率(c)和总皮质锂水平(d)。每个点代表一个个体病例。e,复制队列病例中的总皮质锂。f,锂在MCI和AD的Aβ斑块中富集。AD病例PFC中的Aβ免疫标记(左)。对相邻未固定切片进行激光吸收(LA)-ICP-MS,以量化Aβ斑块(白色圆圈)和邻近非斑块区域(黄色圆圈)中的锂。比例尺,50μm。右侧显示MCI和AD病例中斑块(P)与非斑块(NP)区域的锂水平比率。g,皮质脑样本被亚分馏为富含斑块和非斑块的组分。通过ICP-MS测量非斑块组分中的锂水平,并以NCI的平均值为基准进行标准化。P值通过单因素(a-d)或双因素(f)方差分析与Tukey事后检验和Benjamini-Hochberg校正(a-c)或Tukey事后校正(d、f),或通过双尾未配对t检验(e、g)计算。c-g,箱线图显示个体值、中位数(线)、箱限(第25和第75百分位数)和须线(最小值和最大值)。a-c,NCI n=133,MCI n=58,AD n=94。d,NCI n=177,MCI n=66,AD n=105。e,NCI n=22,AD n=21。f,MCI n=7,AD n=5。g,NCI n=74,AD n=42。
接下来,我们研究了大脑中的内源性锂稳态是否可能受到AD病理的干扰。先前的研究表明,几种金属与Aβ存在相互作用⁸、⁹。为了确定淀粉样蛋白沉积是否影响锂的分布,我们进行了激光吸收(LA)-ICP-MS,并量化了额叶皮层中淀粉样蛋白斑块与无斑块区域的锂含量。在每例MCI和AD病例中,都检测到Aβ斑块中锂的高度显著富集,且从MCI到AD这种富集程度有所增加(图1f)。为了补充这种原位分析,将PFC样本亚分馏为富含斑块的不溶性组分和不含淀粉样蛋白斑块的可溶性组分(补充图1)。与对照NCI病例相比,AD患者PFC非斑块组分中的锂水平平均值和中位数显著降低(图1g)。此外,在整个老年人群中,非斑块皮质组分中较低的锂水平与情景记忆、语义记忆以及整体认知功能指数的认知测试得分降低相关(补充表2)。在AD患者中,非斑块皮质组分中较低的锂水平与情景记忆得分和整体认知功能指数降低相关(补充表2)。
为了进一步探索锂与Aβ的关系,我们检查了内源性锂在J20 Aβ前体蛋白(App)转基因小鼠¹³大脑皮质中的分布,这些小鼠表现出广泛的Aβ沉积。LA-ICP-MS显示,在12月龄J20小鼠的皮质Aβ沉积物中,锂的浓度是相邻无斑块皮质区域的约3-4倍(扩展数据图1d)。此外,皮质的亚分馏显示,与野生型小鼠相比,J20小鼠非斑块皮质组分中的锂显著减少,这与淀粉样蛋白沉积物对锂的封存一致(扩展数据图1e)。相比之下,3月龄J20小鼠在淀粉样蛋白沉积开始前,其可溶性皮质组分中的锂与同龄野生型小鼠相比并未减少(扩展数据图1e)。总之,这些结果表明,锂被Aβ沉积物封存,降低了其生物利用度。
小鼠模型中的锂缺乏
为了探索内源性锂的生物学特性,小鼠被维持在化学成分明确的饮食中,这种饮食在热量和营养上与典型的谷物基小鼠饮食相当,包括相同的锂浓度。食用这种饮食的小鼠的血清和皮质锂水平与老年人群的水平在相似范围内,且平均值无显著差异(见小鼠饮食方法部分)。从小鼠饮食中选择性去除锂(减少92%)导致血清锂平均值降低89%,非斑块组分的皮质锂平均值降低47-52%(扩展数据图2a-c和补充图2)。
在3xTg AD小鼠模型¹⁴(会积累Aβ沉积物和磷酸化tau蛋白)、J20 AD小鼠模型¹³(会积累大量Aβ沉积物)以及无AD型病理的衰老野生型小鼠中,确定了内源性锂减少的病理效应。维持锂缺乏饮食的3xTg和J20小鼠,海马中的Aβ沉积显著增加(图2a、b)。早在食用锂缺乏饮食5周时,就观察到淀粉样蛋白斑块负荷增加,并且随着治疗时间的延长继续增加(图2a、b和扩展数据图2d)。在衰老的野生型小鼠中,锂缺乏饮食使皮质锂减少约50%(扩展数据图2c)。这导致皮质和海马中的Aβ42(AD中的主要致病性Aβ种类)显著升高,Aβ40也有增加的趋势(图2c和扩展数据图2e)。因此,锂缺乏会加速AD小鼠模型中的Aβ沉积,并增加衰老野生型小鼠中的Aβ42水平。
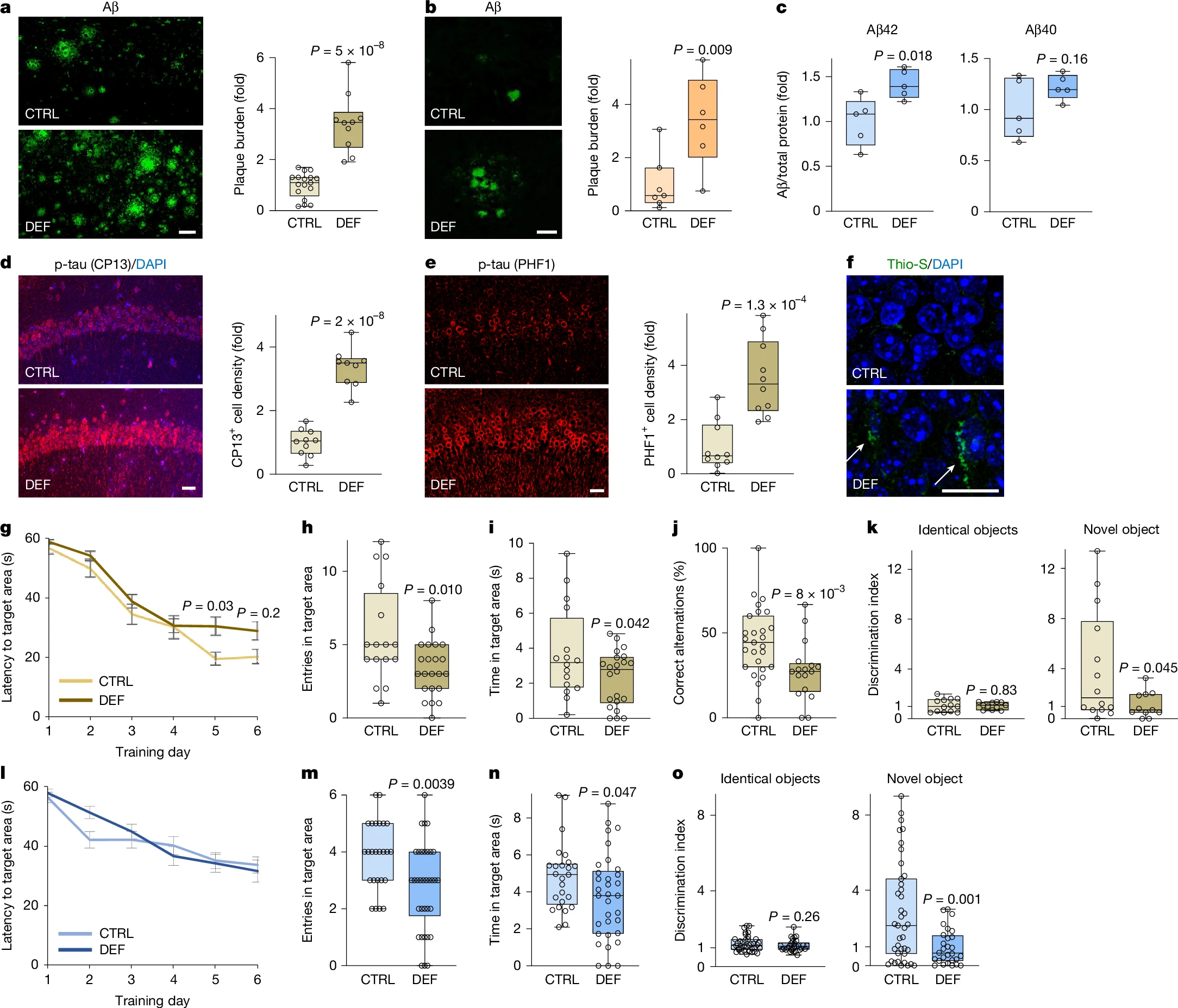 图2:锂缺乏加速AD病理和认知衰退
图2:锂缺乏加速AD病理和认知衰退
a、b,3xTg(a)和J20(b)小鼠在锂缺乏(DEF)或对照(CTRL)饮食下,海马中的Aβ免疫标记和斑块量化(右)。c,野生型(WT)小鼠在DEF或CTRL饮食下,额叶皮层中的Aβ42和Aβ40水平,以总蛋白为基准进行标准化(每组n=5)。d-f,3xTg小鼠在DEF或CTRL饮食下,海马CA1区的pSer202-tau(CP13;d)或pSer396/Ser404-tau(PHF1,e)免疫标记和硫黄素S标记(f),以及磷酸化tau阳性细胞密度的量化(d、e,右)。在f中,箭头指示神经原纤维缠结样结构。g-k,3xTg小鼠在DEF或CTRL饮食下的行为评估:Morris水迷宫学习(g)和记忆(h、i)、Y迷宫(j)和新物体识别(k)。l-o,衰老WT小鼠在DEF或CTRL饮食下的行为评估:Morris水迷宫学习(l)和记忆(m、n),以及新物体识别(o)。3xTg小鼠从6月龄到15月龄(a、d-f)或6月龄到13.5月龄(g-k)接受饮食干预;J20小鼠从3月龄到6月龄(b)接受饮食干预;WT小鼠从12月龄到20月龄(c、l-o)接受饮食干预。在a-e、h-k和m-o中,数据以CTRL为基准进行标准化。在a-e、h-k和m-o中,箱线图显示个体值、中位数(线)、箱限(第25和第75百分位数)和须线(最小值到最大值)。在g和l中,数据为平均值±标准误。P值通过双尾未配对t检验(a-e、h-k、m-o)或混合效应模型与Šídák事后检验(g、l)计算;显示选定的P值。在l中未检测到显著差异。比例尺均为25μm。a,CTRL n=17,DEF n=10;b,CTRL n=7,DEF n=6;d,CTRL n=10,DEF n=9;e,每组n=10;g-i,CTRL n=16,DEF n=22;j,CTRL n=27,DEF n=17;k,CTRL n=13(左),n=14(右);DEF n=11;l-m,CTRL n=25,DEF n=34;n,CTRL n=25,DEF n=33;o,CTRL n=39,DEF n=28。
在3xTg小鼠中,通过评估与AD中神经原纤维缠结(NFT)形成的早期(pSer202-tau)和晚期(pSer396/Ser404-tau)阶段相关的磷酸化tau亚型,探索了锂稳态在tau病理中的作用。在锂缺乏的3xTg小鼠的海马神经元中,pSer202-tau和pSer396/Ser404-tau均增加了3-4倍(图2d、e和扩展数据图2f、g)。部分受影响的神经元在硫黄素S阳性结构中显示出磷酸化tau升高,这些结构类似于NFT(图2f)。与Aβ的观察结果一样,早在锂缺乏饮食5周时就明显出现磷酸化tau升高,并且随着治疗时间的延长继续增加(图2d、e和扩展数据图2f)。这些结果表明,锂缺乏会促进神经元磷酸化tau的积累。
接下来,我们评估了内源性锂在AD型病理存在的情况下以及正常衰老过程中是否影响认知功能。给3xTg小鼠喂食锂缺乏饮食,通过Morris水迷宫实验测定,显著损害了其学习能力(图2g)和长期记忆(图2h、i)。游泳速度、可见平台识别或旷场试验中的表现没有显著变化,这与视觉感知、运动活动和探索行为完好一致(扩展数据图2h-l)。3xTg小鼠的锂缺乏还导致Y迷宫和新物体识别记忆测试的显著缺陷(图2j、k)。此外,衰老的锂缺乏野生型小鼠在Morris水迷宫(图2m、n)和新物体识别测试(图2o)中也表现出显著的记忆丧失。衰老野生型小鼠的这些认知缺陷在空间学习(图2l)、游泳速度、可见平台识别或旷场试验表现无显著变化的情况下出现(扩展数据图2m-q)。因此,内源性锂在AD型病理存在的情况下,以及在小鼠正常衰老过程中,都具有防止记忆丧失的作用。
锂缺乏的转录组
接下来,我们表征了锂缺乏对海马体(MCI和AD中疾病进展的早期部位)细胞群转录组的影响。在给3xTg小鼠喂食锂缺乏饮食5周后,对海马体进行了单核RNA测序(snRNA-seq)分析。我们分别从锂缺乏小鼠和对照小鼠中分析了64,772和54,374个高质量细胞核(补充图3)。基于先前验证的细胞类型特异性标记物的表达(补充图4),在锂缺乏5周后,已解析的细胞类型的相对丰度没有变化(图3a)。
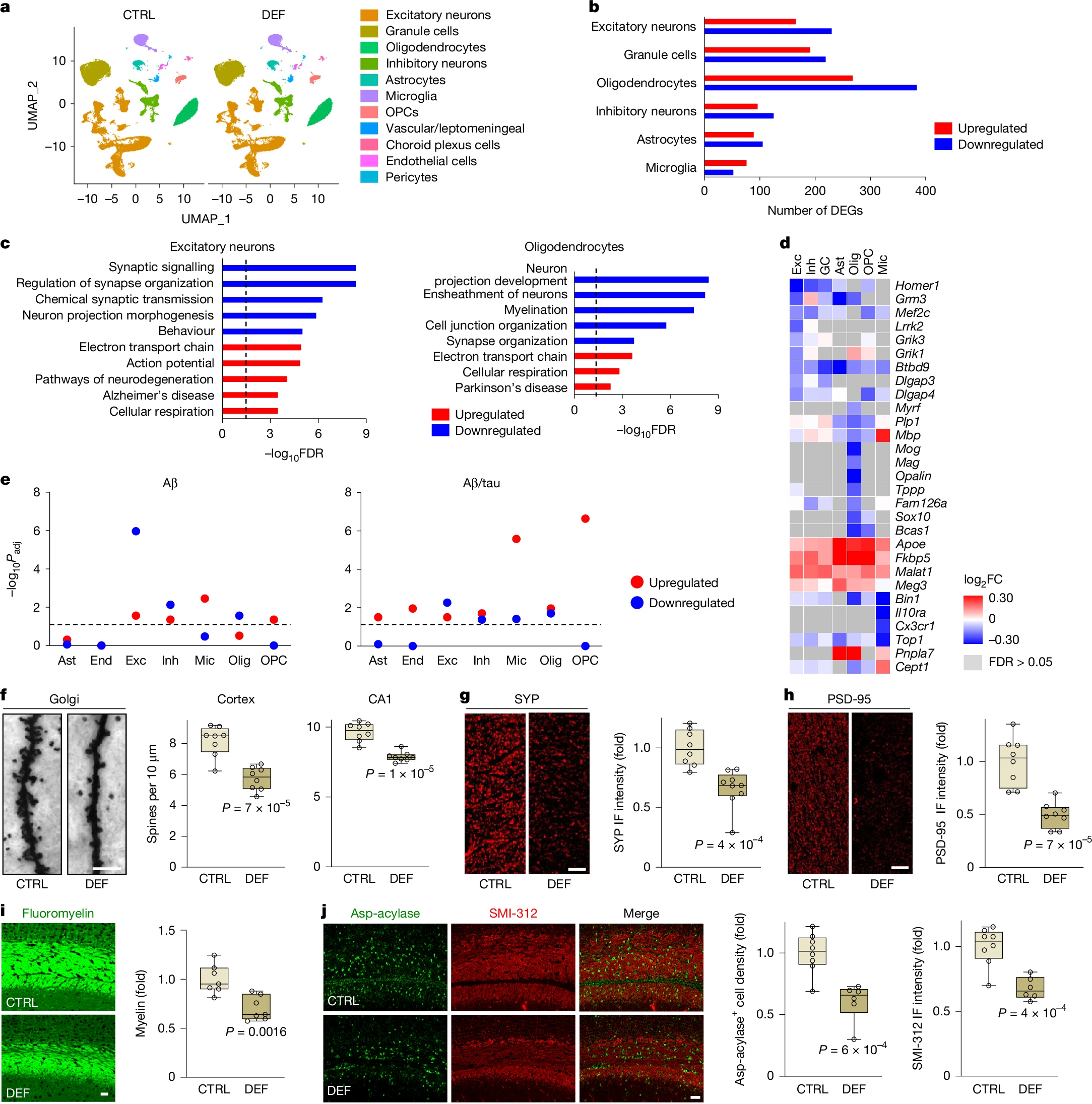
图3:内源性锂对基因表达的细胞类型特异性调控
a. 对接受5周锂缺乏(DEF,n=5)或对照(CTRL,n=4)饮食的12月龄3xTg小鼠海马体进行单核RNA测序(snRNA-seq),得到的细胞核的均匀流形近似和投影(UMAP)图,按细胞类型着色。b. 按方向分层的每种细胞类型的差异表达基因(DEGs)数量。c. 差异表达基因的基因本体(GO)分析,显示富集的下调(蓝色)和上调(红色)通路。d. 热图显示所选差异表达基因在不同细胞类型中的表达变化(log2倍变化)。FC,倍变化。e. 与锂缺乏相关的差异表达基因和人类AD病理相关的差异表达基因的重叠。将接受锂缺乏饮食的3xTg小鼠的snRNA-seq差异表达基因与具有Aβ或Aβ/tau病理的人类活检样本的snRNA-seq差异表达基因进行重叠分析¹⁵。图中显示了每种细胞类型中上调和下调差异表达基因重叠的显著性水平(-log10调整后P值),使用Fisher精确检验计算,并使用Benjamini-Hochberg方法对跨细胞类型和基因方向的多重比较进行校正。f-j. 从6-12月龄(f)或6-15月龄(g-j)接受对照或锂缺乏饮食的3xTg小鼠的突触和结构改变。皮质和海马CA1区的高尔基染色和树突棘密度量化(f)。海马CA1区突触素(SYP;g)和PSD-95(h)的免疫标记和量化;IF,免疫荧光。胼胝体中髓鞘(荧光髓鞘;i)、少突胶质细胞(天冬氨酸酰基转移酶;j)和轴突(SMI-312;j)密度的免疫标记和量化。在f-j中,箱线图显示个体值、中位数(线)、箱限(第25和第75百分位数)和须线(最小值和最大值)。在g-j中,数据以对照组平均值为基准进行标准化。在f-j中,P值通过双尾未配对t检验计算。比例尺:5μm(f)和25μm(g-j)。f、h,每组n=8;g,对照组n=8,锂缺乏组n=9;i,每组n=7;j,对照组n=8,锂缺乏组n=6。Ast,星形胶质细胞;End,内皮细胞;Exc,兴奋性神经元;GC,颗粒细胞;Inh,抑制性神经元;Mic,小胶质细胞;Olig,少突胶质细胞。
对差异表达基因的分析显示,兴奋性神经元、颗粒细胞、抑制性神经元以及少突胶质细胞、星形胶质细胞、小胶质细胞和少突胶质前体细胞(OPCs)中存在显著的转录组变化(图3b以及补充表3和4)。在兴奋性神经元中,与突触信号传导、组织和传递相关的基因本体术语被下调,而电子传递以及神经退行性变和AD相关通路被上调(图3c以及补充表5和6)。下调的突触基因包括Homer1、Grm3、Mef2c、Lrrk2、Grik1、Grik3、Btbd9、Dlgap3和Dlgap4(图3d)。在少突胶质细胞中,与轴突包裹和髓鞘形成以及神经元突起发育相关的基因本体术语被显著下调(图3c以及补充表5和6)。下调的髓鞘相关基因包括Mbp、Mog、Mag、Plp1和Opalin(图3d)。在星形胶质细胞中,神经退行性变、电子传递链和单价阳离子转运相关通路被上调(补充图5以及补充表5和6)。对锂缺乏的3xTg小鼠海马体的蛋白质组学分析显示,突触和髓鞘蛋白成分的丰度显著降低,而参与神经炎症、脂质代谢和线粒体膜组织的蛋白质丰度增加(补充图6和补充表7)。这些结果表明,内源性锂广泛影响大脑转录组和蛋白质组的组成。
接下来,我们通过将我们的发现与最近一项对具有一系列AD病理的活体个体的皮质活检样本进行的snRNA-seq研究进行比较,来研究锂缺乏的转录组是否与AD的转录组重叠¹⁵。人类活检研究中表现出早期Aβ沉积的样本,其差异表达基因与接受锂缺乏饮食的3xTg小鼠存在细胞类型特异性重叠(图3e)。一致性差异表达基因包括兴奋性和抑制性神经元中上调和下调的基因、小胶质细胞和少突胶质前体细胞中上调的基因,以及少突胶质细胞中下调的基因(图3e和补充表8)。在活检前或活检后一年内被诊断为AD且同时具有Aβ和磷酸化tau病理的人类皮质样本中,与锂缺乏差异表达基因的重叠更为广泛¹⁵(图3e)。一致性差异表达基因包括兴奋性和抑制性神经元、小胶质细胞和少突胶质细胞中上调和下调的基因,以及星形胶质细胞、内皮细胞和少突胶质前体细胞中上调的基因(图3e和补充表8)。因此,锂缺乏的转录组与人类AD病理的转录组广泛重叠。
突触和髓鞘的维持
锂缺乏广泛下调了参与突触信号传导和结构的基因(图3c)。通过高尔基染色,在锂缺乏的3xTg和野生型小鼠中检测到树突棘丢失(图3f以及扩展数据图3a、b),同时突触前蛋白突触素和突触后蛋白PSD-95的免疫标记分别减少(图3g、h)。锂缺乏的3xTg海马体的蛋白质组学分析证实了突触蛋白丰度的降低(补充图6a、b和补充表7)。因此,内源性锂有助于衰老小鼠大脑中的突触维持。
锂缺乏下调了少突胶质细胞中髓鞘相关基因的表达,并降低了髓鞘相关蛋白的丰度(图3c、d和补充图6b)。荧光髓鞘标记显示,长期锂缺乏后,3xTg小鼠的髓鞘显著丢失(图3i)。这与锂缺乏的3xTg和野生型小鼠中少突胶质细胞、少突胶质前体细胞和轴突数量的减少相关(图3j以及扩展数据图3c、d)。为了评估髓鞘超微结构,对锂缺乏和对照3xTg小鼠的胼胝体进行了透射电子显微镜检查(扩展数据图3e)。锂缺乏的小鼠围绕神经元轴突的电子致密髓鞘更薄,g值(轴突内径与总轴突加髓鞘纤维直径的比率)显著升高,这与轴突髓鞘减少一致。这些结果表明,内源性锂有助于维持髓鞘的完整性。
锂与小胶质细胞功能
对锂缺乏的snRNA-seq分析显示,表达稳态标志物编码基因Cx3cr1的小胶质细胞数量减少,而表达Apoe的小胶质细胞数量增加(补充图7a),这与AD中观察到的反应性小胶质细胞状态相似¹⁵。为了更深入地了解锂对小胶质细胞的调节作用,从大脑中分离出存活的小胶质细胞,并通过深度RNA测序进行分析。分离的小胶质细胞显示出高度的纯度,并且没有表现出应激相关标志物¹⁶(补充图7b、c)。RNA测序显示,在3xTg和野生型小鼠中,与锂缺乏相关的小胶质细胞存在主要的转录组变化,且重叠程度极高(补充表9-12)。在3xTg和野生型小胶质细胞中,锂缺乏上调的基因在与AD和神经退行性变、电子传递链和呼吸、淀粉样蛋白原纤维形成调节、翻译和氧化应激相关的通路的基因本体术语中富集。锂缺乏下调的基因在DNA损伤反应、细胞对应激的反应、细胞导入和蛋白质分解代谢过程的基因本体术语中富集(图4a、b和补充表12)。
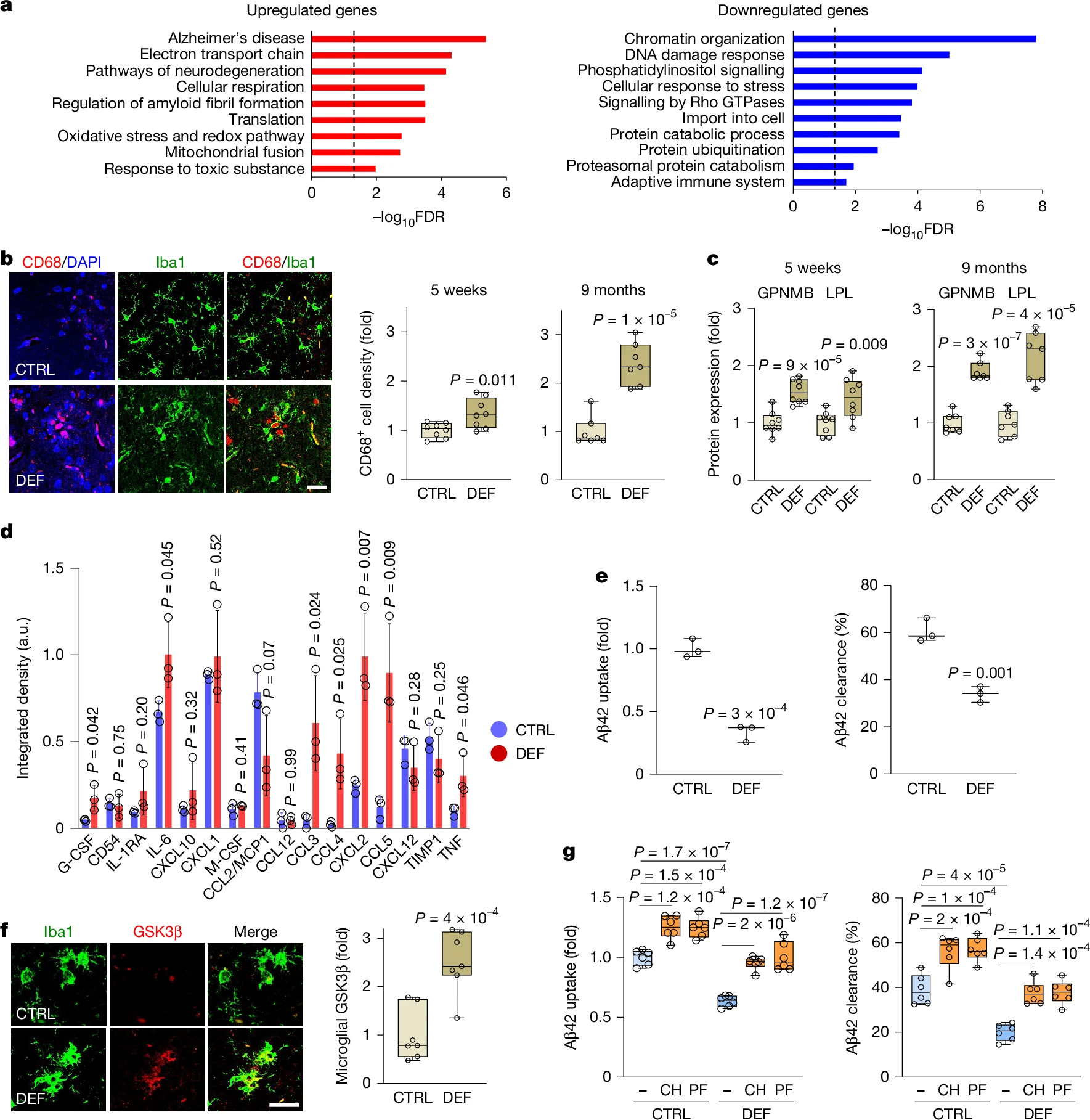
图4:锂缺乏激活小胶质细胞并损害Aβ清除
a. 锂缺乏(DEF)的3xTg小鼠(5至9月龄接受处理;每组n=4只小鼠)和野生型(WT;12至20月龄接受处理;每组n=4只)小胶质细胞之间共有的差异表达基因的GO分析。b、c. 3xTg小鼠接受锂缺乏或对照饮食处理5周(从10.8月龄开始;每组n=8)或9个月(从6月龄开始;每组n=7)。海马体中总小胶质细胞(Iba1)和活化小胶质细胞(CD68)的免疫标记(b,左,9个月处理)。海马体中CD68+细胞密度(b,右)以及GPNMB和LPL小胶质细胞表达(c)的量化。d. 从12至18月龄接受对照或锂缺乏饮食的野生型小鼠中分离出原代皮质小胶质细胞,用50ng/ml脂多糖(LPS)刺激后,培养基中的细胞因子水平(每组n=3)。信号以细胞因子阵列的内部对照为基准进行标准化。数据为平均值±标准差。a.u.,任意单位。e. 18月龄野生型小鼠在接受6个月对照或锂缺乏饮食后,分离出的原代皮质小胶质细胞对Aβ42的摄取(左)和降解(右)(每组n=3)。f. 接受9个月对照或锂缺乏饮食的3xTg小鼠海马体中,Iba1+小胶质细胞中GSK3β的免疫标记(左)和量化(右)(每组n=7)。g. 从12至16月龄接受对照或锂缺乏饮食的野生型小鼠中分离出原代小胶质细胞,在培养中与GSK3β抑制剂CHIR99021(CH)或PF-04802367(PF)共同孵育后,对Aβ42的摄取(左)和降解(右)(每组n=6个生物学重复)。在b-g中,数据以对照组平均值为基准进行标准化。在b、c、e-g中,箱线图显示个体值、中位数(线)、箱限(第25和第75百分位数)和须线(最小值和最大值)。P值通过未配对双尾t检验(a-f)或双因素方差分析与Tukey事后检验(g)计算。比例尺,20μm。
锂缺乏的野生型小胶质细胞的转录组变化在全基因组关联研究(GWAS)中确定的许多AD风险基因中富集¹⁷,包括Apoe、Trem2、Bin1、Clu、Picalm、Cd33、H2-Eb1(HLA-DRB1同源物)、Inpp5d、Abca1、Abca7和Adam10(基因组注释的多标记分析(MAGMA),错误发现率(FDR)<0.05)。我们还观察到锂缺乏小胶质细胞的转录组特征与AD中小胶质细胞的转录组特征显著重叠,特别是对于表达糖蛋白NMB(GPNMB)的小胶质细胞(3xTg小鼠P<10⁻²¹,野生型小鼠P<10⁻¹⁴),其随着AD的进展而扩增¹⁵。此外,GPNMB表达在锂缺乏的小胶质细胞中显著上调(补充表11)。因此,锂缺乏以与AD重叠的方式改变小胶质细胞的转录组。
接下来,我们研究了锂缺乏后小胶质细胞的反应性变化。免疫标记显示,锂缺乏增加了3xTg小鼠中CD68免疫反应性小胶质细胞(图4b),并提高了小胶质细胞中GPNMB和脂蛋白脂肪酶(LPL)的蛋白表达,这些是AD中小胶质细胞反应性的标志物¹⁵(图4c)。在第二种转基因AD小鼠模型J20中,锂缺乏也增加了CD68+反应性小胶质细胞的密度(补充图7d)。为了探索功能变化,在培养中用脂多糖(LPS)刺激分离出的小胶质细胞。来自锂缺乏的野生型小鼠的小胶质细胞显示促炎细胞因子IL-6、TNF和G-CSF以及免疫激活趋化因子CCL3、CCL4、CCL5和CXCL2的释放增加(图4d)。最后,与来自对照小鼠的小胶质细胞相比,从锂缺乏的野生型小鼠中分离出的小胶质细胞对Aβ42的摄取和随后的降解显著减少(图4e)。因此,锂缺乏导致反应性促炎状态和Aβ清除受损。
内源性锂对GSK3β的调节
接下来,我们使用差异表达基因的Ingenuity/IPA网络分析来确定受锂缺乏影响的信号通路。Wnt-β-连环蛋白信号传导受到显著影响,并且预计在小胶质细胞、兴奋性神经元和少突胶质细胞中受到抑制(扩展数据图4a-c)。免疫标记显示,在锂缺乏的3xTg和野生型小鼠的海马CA1神经元中,核β-连环蛋白水平显著降低(扩展数据图5a、b)。在锂缺乏的少突胶质细胞和小胶质细胞中,核β-连环蛋白水平也降低(扩展数据图5c、d)。
β-连环蛋白信号传导的一个核心调节因子是丝氨酸-苏氨酸激酶GSK3β,它使β-连环蛋白磷酸化,使其靶向蛋白酶体降解。另一种GSK3β底物是tau蛋白,在AD中GSK3β使tau蛋白磷酸化¹⁸、¹⁹、²⁰、²¹,并且在锂缺乏的3xTg小鼠中显示出3-4倍的磷酸化升高(图2d、e)。锂缺乏提高了3xTg小鼠海马CA1神经元、少突胶质细胞和小胶质细胞中的总GSK3β水平(图4f以及扩展数据图5e、f)。锂缺乏小鼠海马体的蛋白质组学分析证实了总GSK3β蛋白的升高(补充表7)。锂缺乏也使GSK3βmRNA升高(扩展数据图5g),这与GSK3β表达增加一致。
GSK3β通过酪氨酸216的自磷酸化被激活,这在AD中有所增加¹⁸、²¹。在锂缺乏的3xTg和野生型小鼠的海马CA1神经元和少突胶质细胞中,pTyr216-GSK3β水平显著升高(扩展数据图5h、i)。Ser9处GSK3β的磷酸化(抑制GSK3β活性)在绝对水平上没有变化,但pSer9:总GSK3β的比率降低,这与GSK3β活性升高一致(扩展数据图5j、k)。相比之下,肌醇的水平(锂在药理浓度下通过肌醇单磷酸酶调节²²)不受内源性锂缺乏的影响(扩展数据图5l)。
为了评估GSK3β在锂缺乏的致病效应中的作用,向锂缺乏的3xTg小鼠施用GSK3β抑制剂CHIR99021。CHIR99021处理逆转了与锂缺乏相关的小胶质细胞激活(扩展数据图6a),并使多种促炎细胞因子和趋化因子的水平正常化(扩展数据图6b)。此外,将来自锂缺乏的野生型小鼠的原代小胶质细胞与CHIR99021共同孵育,恢复了Aβ42的摄取和降解(图4g)。使用第二种GSK3β抑制剂PF-04802367也获得了类似的结果(图4g)。此外,用CHIR99021处理锂缺乏的3xTg小鼠,逆转了升高的Aβ沉积和tau磷酸化,并恢复了少突胶质细胞数量和髓鞘碱性蛋白(MBP)的表达(扩展数据图6c-e)。因此,GSK3β激活促成了与锂缺乏相关的广泛病理。
锂替代疗法
观察到锂在MCI和AD中被淀粉样蛋白沉积物封存,促使人们寻找与淀粉样蛋白结合减少的治疗性锂盐。我们推断,锂离子与Aβ沉积物的静电相互作用是盐的电离能力的函数,而电离减少的锂盐可能显示出淀粉样蛋白封存减少。为了直接评估电离,我们测量了16种锂盐的电导率。无机锂盐,包括临床标准碳酸锂(Li₂CO₃,以下简称LiC),与有机锂盐相比,显示出显著升高的电导率,表明电离增加(P=8×10⁻⁴;图5a和扩展数据图7a)。在有机锂盐中,乳清酸锂(C₅H₃LiN₂O₄,以下简称LiO)在广泛的锂浓度范围内显示出最低的电导率(图5a和扩展数据图7a),因此被选择用于与临床标准LiC进一步比较。
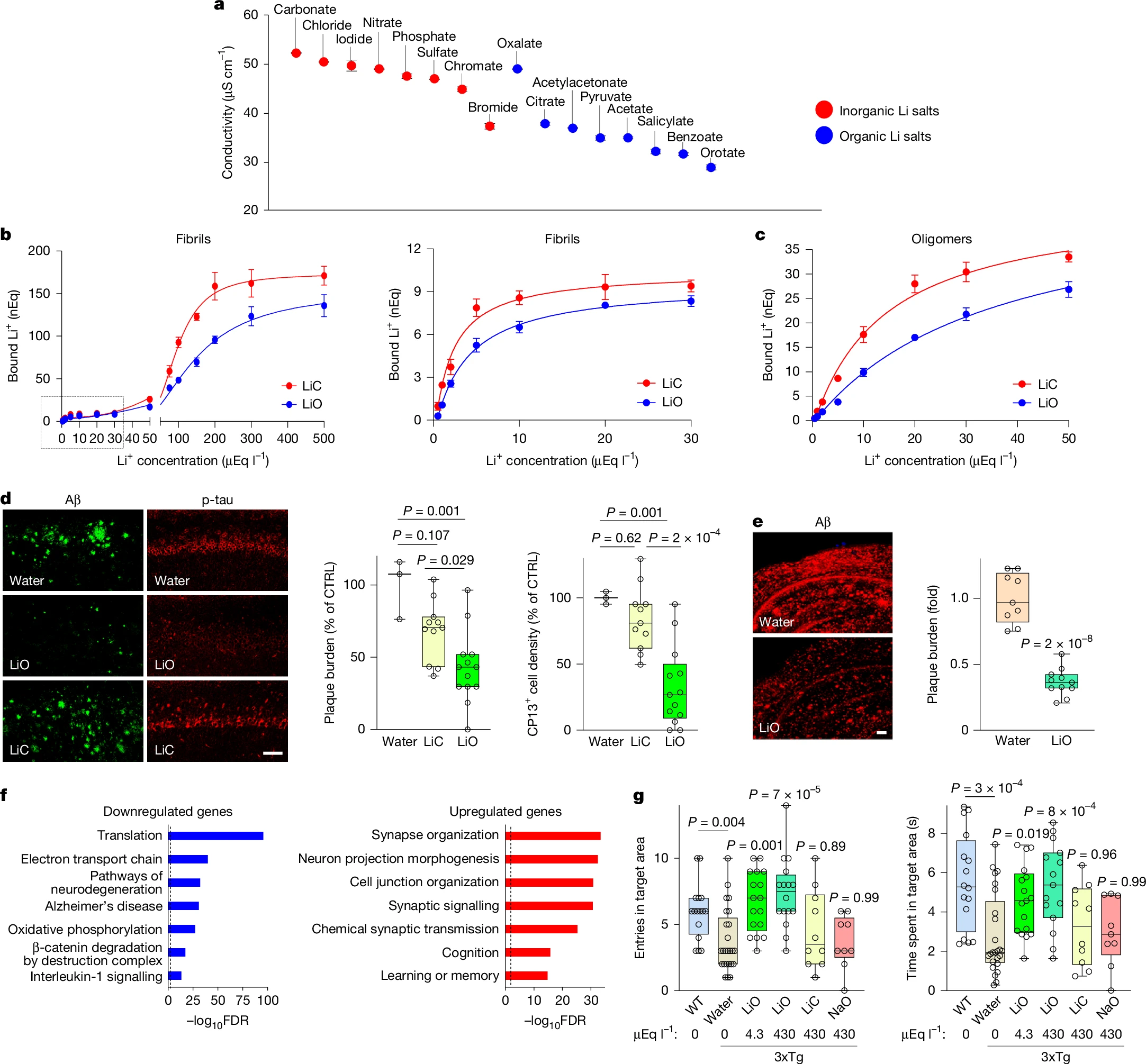
图5:一种可避开斑块的锂盐的治疗效果
a. 有机锂盐溶液的电导率低于无机锂盐。所有溶液均含有430µEq/l锂。b、c. 锂与人Aβ1-42原纤维(b)和寡聚物(c)结合。显示所有测试浓度的结合曲线(b,左)和0-30µEq/l范围的结合曲线(b,右)。LiC显示出比LiO更高的亲和力(补充表13中为EC50值和95%置信区间)。d. 从9至18月龄接受LiO或LiC(4.3µEq/l)处理的3xTg小鼠海马体中Aβ和pSer202-tau的免疫标记(左)以及斑块负荷(中)和pSer202-tau+细胞密度(右)的量化(溶媒n=3,LiC n=11,LiO n=13)。e. 从17至22月龄接受LiO(4.3µEq/l)处理的J20小鼠中Aβ的免疫标记(左)和量化(右)(水n=9,LiO n
接下来,我们研究了电导率是否能预测锂与阿尔茨海默病(AD)中积累的β-淀粉样蛋白(Aβ)聚集形式的结合。平衡透析结合实验表明,在体外,锂会与由合成的人Aβ1-42形成的原纤维和寡聚物结合(图5b、c以及扩展数据图7b)。锂的结合具有饱和性,其高亲和力范围包括大脑和血清中锂的生理浓度范围(0-2µEq/l),而低亲和力范围则对应临床药物剂量。值得注意的是,在较广的锂浓度范围内,碳酸锂(LiC)对Aβ42原纤维和寡聚物的结合亲和力均高于乳清酸锂(LiO)(图5b、c以及补充表13)。
为了进一步在体内探索锂与Aβ的相互作用,我们以低剂量在饮用水中给J20和3xTg小鼠施用LiO和LiC(4.3μEq/l锂),使得血清锂水平处于在衰老人类和小鼠中观察到的生理浓度范围内(补充图8a、c、d、扩展数据图1c及方法部分)。施用低剂量LiO或LiC后,血清和海马中的锂水平无显著差异(补充图8a、b)。然而,在3xTg和J20小鼠中,施用LiC后,锂在Aβ斑块中高度富集,而施用LiO后,这种富集程度要低得多(扩展数据图7c、d)。此外,在3xTg和J20小鼠中,LiO(而非LiC)显著提高了非斑块部分的脑实质锂水平(扩展数据图7e、f)。因此,与LiC相比,LiO的淀粉样蛋白封存作用减弱,能更有效地提高大脑中非斑块部分的锂水平。
给成年3xTg小鼠施用LiO时,生理剂量几乎能完全阻止Aβ斑块沉积和磷酸化tau蛋白积累;相比之下,施用LiC或乳清酸钠对照则无显著效果(扩展数据图8a、b)。接下来,我们研究了LiO是否能逆转衰老小鼠中更严重的病理状况。从9到18月龄给3xTg小鼠施用LiO,显著降低了海马中Aβ斑块负荷以及磷酸化tau阳性的神经原纤维缠结样结构的密度,而相同剂量的LiC则无显著效果(图5d)。在具有大量且广泛Aβ沉积的老年J20小鼠中,LiO还能使Aβ斑块负荷减少约70%(图5e)。因此,LiO在减少Aβ沉积和磷酸化tau积累方面效果显著。
LiO(而非LiC)能增加突触标志物PSD-95的表达(扩展数据图8c),提高髓鞘相关髓鞘碱性蛋白(MBP)的免疫反应性,并增加少突胶质细胞数量(扩展数据图8d、e)。在抑制老年3xTg小鼠的小胶质细胞增生和星形胶质细胞增生方面,LiO也比LiC更有效(扩展数据图8f、g)。施用低剂量LiO(而非LiC)还能降低海马CA1神经元和白质中的总糖原合成酶激酶3β(GSK3β)水平(扩展数据图9a、b),减少活化的pTyr216-GSK3β水平(扩展数据图9c),并提高核β-连环蛋白水平(扩展数据图9d)。
对接受LiO处理的3xTg小鼠海马体进行RNA测序分析显示,与翻译、电子传递链、神经退行性变通路、阿尔茨海默病、β-连环蛋白降解和白细胞介素-1信号传导相关的基因本体术语对应的基因表达下调,而与突触组织和信号传导、神经元突起形态发生以及学习或记忆相关的基因本体术语对应的基因表达上调(图5f以及补充表14和15)。这些基因本体术语与锂缺乏相关的术语相似,但变化方向相反(图3c和图4a)。
LiO对参与学习和记忆的基因产生影响,这促使我们探索3xTg小鼠的认知功能,这些小鼠与野生型小鼠相比存在记忆丧失(图5g)。最低剂量的LiO(4.3μEq/l)几乎能完全逆转记忆丧失,而LiC和乳清酸钠对照则无显著效果(图5g)。此外,LiO能改善具有严重淀粉样蛋白病理的衰老J20小鼠的学习和空间记忆(扩展数据图10a-c)。LiO对游泳速度、视觉平台定位或旷场试验中的运动表现无显著影响(扩展数据图10d-h以及补充图9a-f)。因此,LiO能抑制AD型病理、神经炎症和突触丢失,并恢复记忆。
锂与大脑衰老
为了探索锂对正常大脑衰老的影响,我们从12到24月龄给野生型小鼠施用低剂量LiO(4.3μEq/l锂)。这使得血清和皮质锂水平的平均值适度升高,与未处理小鼠的内源性锂范围重叠(补充图10a)。LiO几乎能完全阻止海马体、皮质和胼胝体中与年龄相关的小胶质细胞增生和星形胶质细胞增生(扩展数据图11a、b)。此外,LiO减少了与年龄相关的促炎细胞因子IL-6和IL-1β的产生(扩展数据图12a)。
与来自年轻成年小鼠的小胶质细胞相比,从老年小鼠中分离出的小胶质细胞降解Aβ42的能力显著降低。在体内用LiO处理可逆转这种与年龄相关的Aβ降解能力丧失(扩展数据图12b)。在培养中用LiO孵育BV2小胶质细胞也得到了类似结果。LiO显著提高了BV2细胞对Aβ42的摄取和降解,而对照乳清酸钠则无显著效果(扩展数据图12c-e)。因此,LiO能减少与年龄相关的神经炎症变化,并恢复小胶质细胞清除Aβ的能力。
突触丢失和认知衰退是小鼠大脑衰老的特征。给野生型小鼠施用LiO可防止CA1和CA3海马神经元中与年龄相关的树突棘丢失,而对照盐NaO则无显著效果(补充图10b)。重要的是,通过Morris水迷宫试验确定,LiO在很大程度上逆转了与衰老相关的学习和记忆衰退(扩展数据图13a、b)。LiO对游泳速度或可见平台的定位无影响(扩展数据图13c、d)。LiO还能防止与年龄相关的新物体识别记忆衰退(扩展数据图13e)。值得注意的是,给衰老的野生型、3xTg或J20小鼠长期施用LiO,并未改变血清中血尿素氮、肌酐或促甲状腺激素(TSH)的水平(补充图11)。因此,低剂量LiO能防止小鼠与年龄相关的促炎变化、突触丢失和认知衰退,且无毒性迹象。
这些观察结果促使我们研究正常衰老人类的内源性大脑锂水平与认知弹性的关系。调节突触囊泡胞吐作用的突触前蛋白complexin 1和2的表达水平可预测对AD的抵抗能力²³、²⁴。在没有轻度认知障碍(MCI)或AD的老年人中,complexin 1和2的表达与锂的皮质与血清比率呈高度显著的正相关(扩展数据图13f)。此外,皮质锂水平与工作记忆的认知测试得分(P=0.04)以及简易精神状态检查(MMSE)得分(P=0.02)呈正相关。总之,这些来自正常衰老小鼠和人类的结果表明,锂稳态可能有助于认知弹性。
讨论
这些观察结果表明,内源性锂具有影响大脑衰老和AD易感性的生理作用。在人类中,锂在精神病学中用于治疗双相情感障碍,其剂量范围会使血清中的锂水平升高到内源性水平的约1000倍。我们发现,在正常衰老的小鼠中,低微摩尔水平的内源性锂可维持认知功能、减少炎症并抑制Aβ生成。在AD小鼠模型中,内源性锂能防止淀粉样蛋白沉积、tau蛋白过度磷酸化、神经炎症以及突触、轴突和髓鞘的丢失。这些效应至少部分是通过抑制大脑中多种细胞类型中的激酶GSK3β介导的。
大脑的金属组学分析还发现了其他金属的变化,这些变化先前在AD中也有观察到,例如钠²⁵和锌²⁶的增加以及铜¹²的减少。然而,在我们的数据集中,皮质锂的减少是在MCI和AD中均观察到的唯一具有统计学意义的金属变化。这一观察结果与丹麦的一项人群研究一致,该研究发现当地饮用水中的锂水平与痴呆症发病率呈显著的负相关²⁷。我们能够通过锂缺乏饮食在小鼠模型中重现AD患者中观察到的锂水平降低。单核RNA测序显示,皮质锂水平降低会显著改变主要脑细胞类型的转录组。此外,小鼠模型中与锂缺乏相关的差异表达基因,与先前在具有AD病理的人类脑活检样本的单核RNA测序中鉴定出的差异表达基因存在重叠¹⁵。重叠的差异表达基因在小胶质细胞、兴奋性和抑制性神经元、少突胶质前体细胞、少突胶质细胞、内皮细胞和星形胶质细胞中富集。与这些转录组变化一致,锂缺乏对神经元、小胶质细胞和少突胶质细胞产生有害影响,导致突触和轴突丢失、Aβ清除受损以及髓鞘形成减少。
锂缺乏改变了小胶质细胞中一些在AD全基因组关联研究中确定的最具渗透性的风险因子基因的表达,包括Apoe、Trem2、Bin1、Picalm、Clu、Cd33以及人类MS4A6A的小鼠同源物¹⁷。这些表达变化与小胶质细胞活化标志物(如CD68)的升高以及多种促炎细胞因子和趋化因子的产生增加相关,这些在AD中也会升高²⁸。此外,锂缺乏会损害小胶质细胞吞噬和降解Aβ的能力,这一表型与AD中的Aβ沉积有关²⁹。这些发现表明,内源性锂可维持小胶质细胞的稳态功能,并防止与AD相关的促炎变化。
在药物剂量下,锂的首批被表征的分子靶点之一是激酶GSK3β³⁰。GSK3β活性在AD中会增强,并与Aβ和tau病理有关¹⁸、²⁰、²¹、³¹。此外,AD中GSK3β的底物β-连环蛋白会减少¹⁸、³²、³³。据报道,体外锂与GSK3β相互作用的半数抑制浓度(IC50)在毫摩尔范围内³⁰,这使得在大脑内源性锂的低微摩尔浓度范围内,这种相互作用看似不太可能发生。然而,我们的发现表明,内源性锂缺乏会导致GSK3β活化和表达升高。此外,GSK3β抑制剂可逆转锂缺乏的许多病理后果,包括Aβ沉积、磷酸化tau积累、髓鞘形成和小胶质细胞促炎活化,以及恢复小胶质细胞清除Aβ的能力。先前的报道表明,GSK3β活化可促进单核细胞和小胶质细胞中细胞因子的产生³⁴,并通过抑制β-连环蛋白信号传导损害少突胶质细胞分化和髓鞘形成³⁵。总之,我们的发现表明,GSK3β活性增加与大脑中锂缺乏的多系统效应有关。
已有研究表明,在药物剂量下施用锂,可减少各种神经退行性疾病模型中的Aβ³³、³⁶、³⁷、³⁸、³⁹、⁴⁰、⁴¹、⁴²、⁴³、⁴⁴、⁴⁵和tau³⁷、³⁸、⁴³、⁴⁶、⁴⁷、⁴⁸、⁴⁹病理。同样,在几项研究中,锂可防止或逆转认知衰退³⁶、³⁷、³⁸、³⁹、⁴³、⁴⁴,但在一项短期研究中则不能⁴⁷。几项小型临床试验为锂在AD中的治疗作用提供了更直接的证据。在两项初步试验⁵⁰、⁵¹中,锂治疗并未改善认知功能,但在随后的三项试验中,较低浓度的锂(血清中0.25-0.5mEq/l)减少了认知衰退⁵²、⁵³、⁵⁴、⁵⁵。这些临床试验的一个局限性可能是使用了具有高淀粉样蛋白结合水平的锂盐。我们表征了一种锂盐——乳清酸锂,其结合力降低,能够绕过AD小鼠模型中的淀粉样蛋白封存。以维持血清和皮质锂水平在内源性范围内的剂量施用LiO,可防止和逆转AD小鼠模型和衰老野生型小鼠中的AD病理、神经炎症变化和记忆丧失。在老年个体中使用药物剂量的锂进行治疗时,一个重要的局限性是肾脏和甲状腺毒性⁵⁶。令人鼓舞的是,给衰老小鼠长期施用低剂量LiO后,未检测到毒性。
锂稳态的破坏可能会导致在临床AD发作之前出现的长期前驱期神经病理变化。我们的发现表明,淀粉样蛋白对锂的封存会耗尽受影响脑区的锂。反过来,锂耗竭会损害小胶质细胞对Aβ的清除,这将通过正反馈循环加速淀粉样蛋白病理。同时,锂缺乏可能会促进磷酸化tau积累、炎症以及突触、轴突和髓鞘的丢失。这种神经退行性过程的进展可能受到遗传风险变异¹⁷、⁵⁷以及环境因素和饮食锂摄入的调节。因此,锂缺乏是导致大脑多系统退化并引发AD的潜在共同机制。
https://wap.sciencenet.cn/blog-41174-1496911.html
上一篇:50年前彻底改变生命科学与医疗健康领域的单抗技术【自然】
下一篇:氢医学的现状、挑战与未来方向【何前军】