博文
Phenomics | 中山大学贺雄雷教授团队发布表型空间编码规则
|
近日,《表型组学(英文)》(Phenomics)在线发表了中山大学贺雄雷教授团队题为“The trait coding rule in phenotype space”的研究论文。
研究团队开发了一种基于维度分解的方法(UBHDD),发现表型空间由有限的遗传决定维度(PG)和无限的非遗传塑造维度(PNG)共同编码,这一规则在酵母及人类大脑表型组数据中均得到验证,揭示了遗传因素主导共性维度而非遗传因素驱动特异维度的表型编码规律,为新兴的表型组学领域建立了一个基础理论。
文末点击“阅读原文”可在线阅读文章。

扫描二维码 | 下载PDF原文
论文DOI链接:
https://doi.org/10.1007/s43657-024-00195-5
论文引用格式:
Wang, J., He, X. The Trait Coding Rule in Phenotype Space. Phenomics (2025). https://doi.org/10.1007/s43657-024-00195-5
研究背景
现代生物学长期以基因型为中心解释表型,遵循自下而上的还原论范式,但这一路径仅在简单表型中取得有限成功。类似物理学中宏观与微观的双重描述框架,表型作为宏观层可能蕴含独立于基因型的编码规律。尽管已有研究揭示了表型维度间的共享、协调与权衡现象,但这些发现因局限于特定性状和物种,尚未形成普适性理论框架。近年来,多物种大规模表型组数据的积累(如酵母、人类脑表型库),为探索表型层的统一规律提供了新契机。定量遗传学将表型解构为遗传(PG)与非遗传(PNG)子空间,Fisher的适应性几何模型进一步预测:PG维度受“复杂性成本”限制而高度精简,而PNG因环境随机性、发育可塑性等因素趋向无限维度。这一理论暗示,解析表型空间需优先分离PG与PNG,但现有方法依赖基因型数据且无法直接刻画高维非遗传噪声。如何基于表型组数据本身实现子空间分离,成为揭示遗传编码共性与环境塑造特异性的关键挑战,亦为该研究的核心出发点。
针对表型空间遗传与非遗传子空间分离的难题,该研究提出了一种基于统计独立性的数学框架——通过分析多维表型间边际相关性与高维依赖关系的解耦规律,开发出基于非相关性的高维依赖理论(UBHDD)。该方法无需依赖先验基因型信息,直接利用表型数据自身的几何特性,将PG与PNG分离:研究团队在酵母(约400性状×约1000个体)和人类脑表型组(约700性状×约26000个体)中验证了该方法的有效性。该方法进一步应用于人脑表型分析,首次量化了脑左右不对称性的遗传贡献比例,并挖掘出数百个脑区与精神疾病的新型遗传关联。
研究结果
基于非相关性的高维依赖理论(UBHDD)
研究通过几何空间分析揭示:向量间的高维依赖源于维度共享而非边际相关性(图1a-c)。推导发现,在有限维度空间中,强相关向量大概率共享维度;而低相关向量在低维空间仍可能共享维度,但在高维空间共享概率趋近于零。基于此,研究提出UBHDD方法——通过筛选与目标表型不相关但共享遗传维度的表型,构建仅捕获遗传成分的预测模型,其残差即为非遗传成分。该方法利用PG维度有限而PNG维度无限的特性,通过维度解偶实现表型空间的遗传/非遗传子空间分离(图1e)。
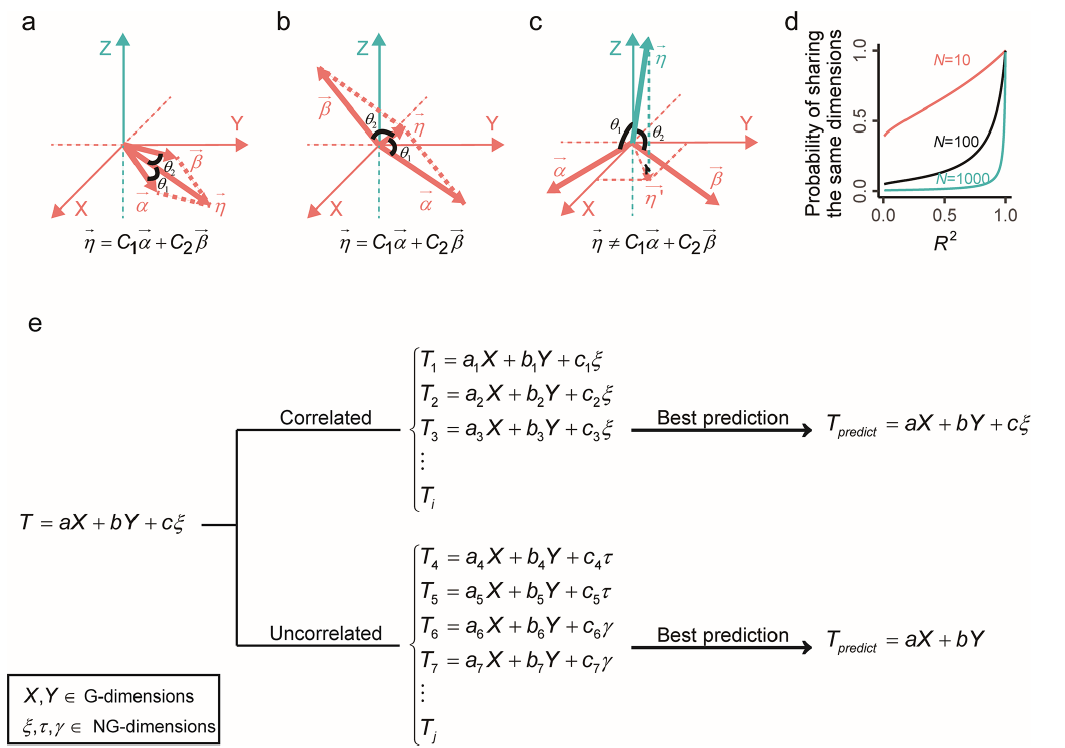
图1 UBHDD的数学理论
UBHDD方法的模拟验证
为检验UBHDD分离不同维度子空间的能力,研究构建模拟表型空间P:其遗传子空间PG仅含10个潜在维度,非遗传子空间PNG包含10000个高维噪声。通过随机线性组合生成1000个表型性状,每个性状的遗传成分方差等于广义遗传率(H²)。应用UBHDD后,发现PG的维度随性状采样迅速饱和,而PNG维度持续增长,符合“有限遗传维度+无限非遗传维度”的理论假设(图2a)。分析表明,UBHDD预测的遗传成分与实际高度吻合,且方差能准确反映性状的遗传力(图2c-d)。进一步比较显示,UBHDD与主成分分析(PCA)存在本质差异:UBHDD是维度解耦而非降维。在模拟存在强相关性状集群的结构化数据中,UBHDD不受干扰仍能准确分离PG与PNG(图2f);而PCA因过度拟合集群结构,无法区分遗传与非遗传信号(图2g)。这表明UBHDD的分离能力源于其基于统计独立性的数学框架,而非依赖数据分布假设。
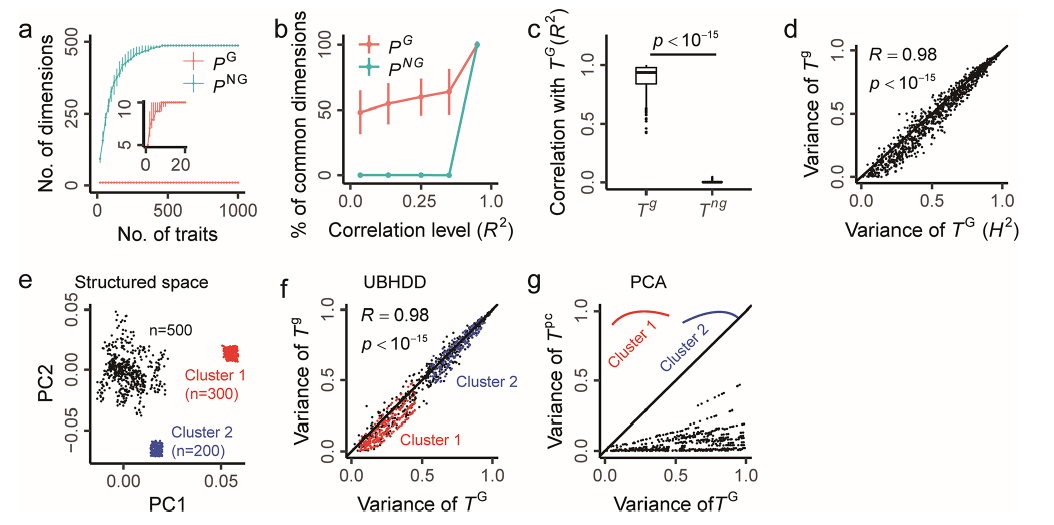
图2 UBHDD在模拟表型空间中分离遗传与非遗传成分
UBHDD在酵母表型空间中的应用验证
研究基于815株酵母的405个形态表型数据(涵盖细胞形态、核定位等性状,广义遗传率H²中位数0.42),利用UBHDD分离遗传与非遗传成分。结果显示,遗传成分解释的性状方差中位数为0.53(图3d),且通过随机置换实验排除了技术偏差干扰(置换后最大方差仅0.013)。与基于重复样本的线性混合模型(LMM)结果对比发现,UBHDD预测的遗传成分与实际遗传成分高度一致(图3e-f),其方差与LMM估计的H²匹配(图3g)。关键验证表明,遗传成分的狭义遗传率(h²)及QTL检测数量显著高于非遗传成分(图3h-i),证实UBHDD有效捕获遗传信号。研究同时指出局限性:样本量限制可能导致遗传成分估计偏差;性状多样性不足或使部分非遗传维度混入遗传成分;LMM因仅两个重复也存在误差。但整体表明,UBHDD无需基因型数据即可从表型组中解析遗传架构,为复杂性状分析提供了新工具。研究进一步在基因缺失酵母群体(del-population,含约5000株非必需基因缺失菌株)中应用UBHDD,分离出各性状的遗传成分。将其与分离群体(seg-population)的UBHDD结果对比发现:两个群体学习到的遗传成分函数高度一致。例如,性状C11.1_A在del群体中预测的遗传成分与seg群体模型所得遗传成分相似度达0.88(图3k);405个性状的相似度中位数为0.82(图3l)。这表明UBHDD解析的遗传子空间(PG)具有跨群体稳健性。
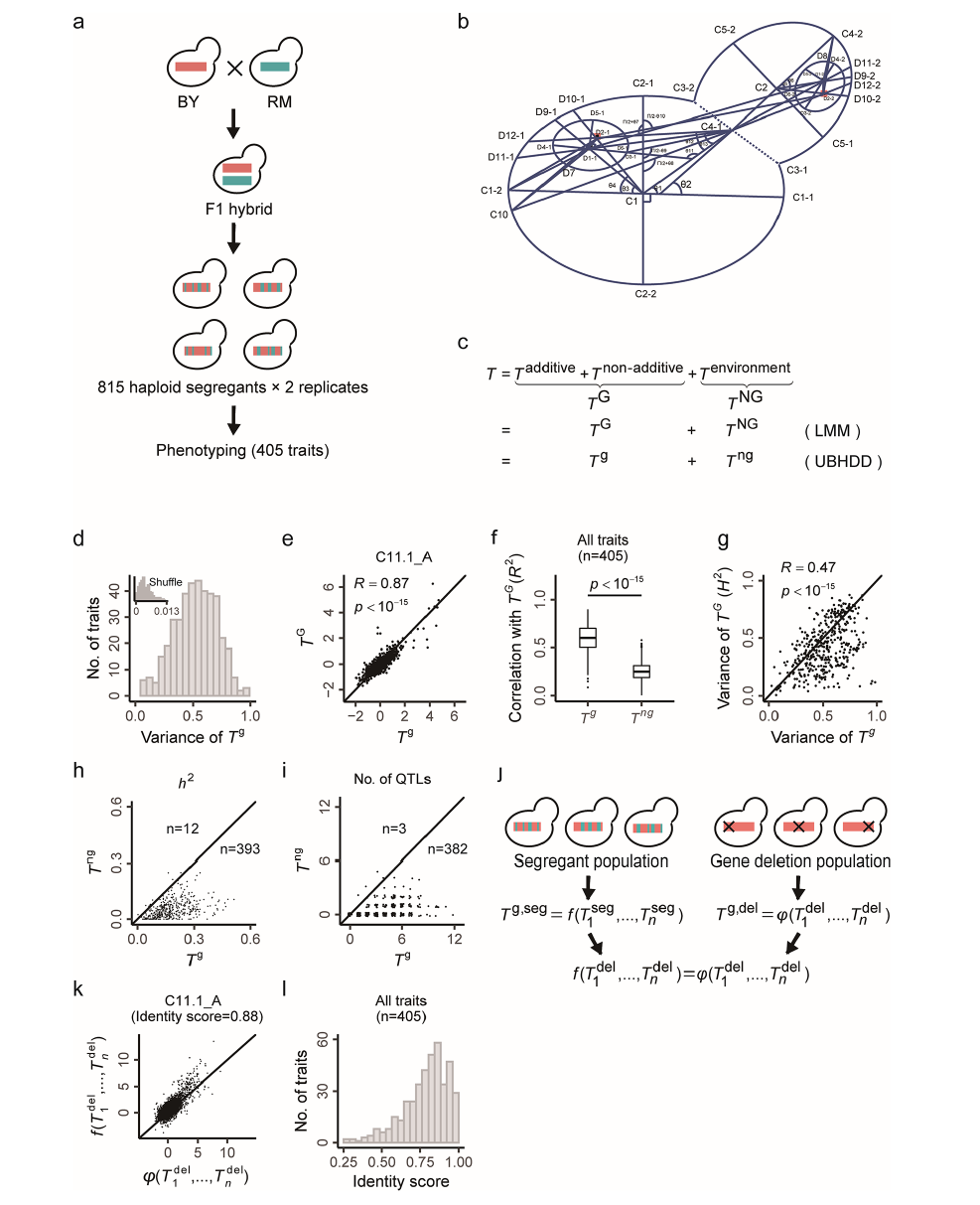
图3 UBHDD实现的405个酵母性状遗传和非遗传成分分离
UBHDD解析人类脑表型空间的遗传架构
研究基于英国生物银行(UK Biobank)25,957名个体的675个脑部影像表型,应用UBHDD分离遗传与非遗传成分。结果显示,遗传成分解释的性状方差中位数为0.48(图4b),且通过置换实验排除技术干扰(置换后最大方差仅4e-4)。与酵母结果一致,遗传成分的狭义遗传率(h²)及QTL数量显著高于非遗传成分(图4c-d),证实遗传成分富集遗传信号。通过分析297对对称脑区表型发现,左右脑不对称性主要由非遗传因素驱动——对称脑区遗传成分的个体间相关性(R²)显著高于非遗传成分(图4e),提示环境或发育随机性主导脑结构偏侧化。基于遗传成分的遗传相关性分析,发现:(i)脑区特异性:如“额枕上束”与19种精神疾病/认知特征(如抑郁症、教育程度)相关,而“穹窿”仅与精神分裂症关联(图5d);(ii)疾病异质性:28种精神疾病中,13种与“海马旁回扣带束”遗传相关,而13种呼吸/循环系统疾病仅2种关联该区域;(iii)左右脑分化:创伤后应激障碍、冠心病等疾病呈现显著单侧脑区关联,而睡眠时长、高血压则双侧均衡(图5e)。研究新发现数百个脑区-疾病关联,如“小脑中脚”的五项指标同时与呼吸困难、高血压遗传相关;“额枕上束”与抑郁症、“小脑上脚”与大麻使用障碍的关联强度超越既往研究。这些发现为解析精神疾病脑机制提供了全新线索。
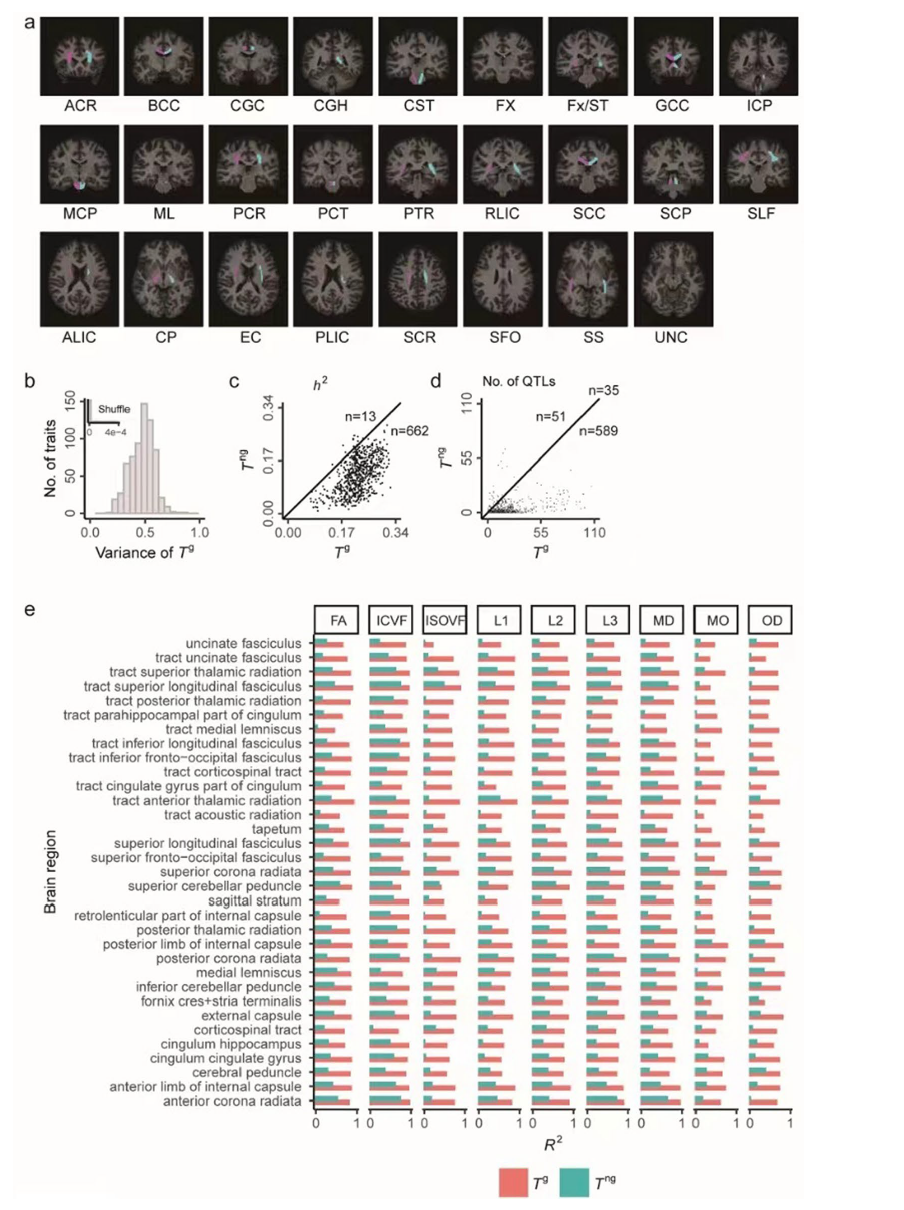
图4 基于UBHDD的人类脑影像表型遗传子空间解析
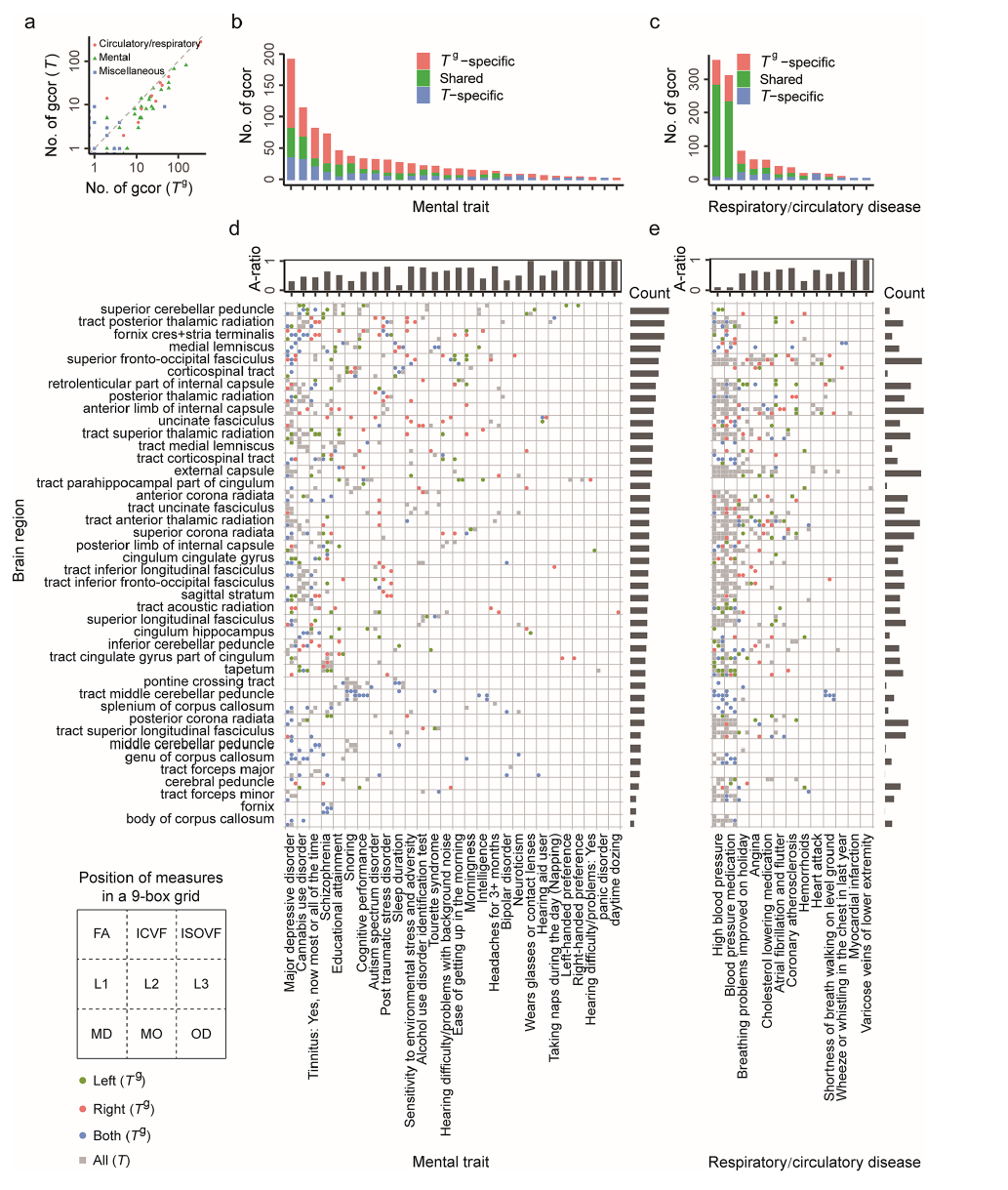
图5 脑区影像性状与精神性状/疾病的新遗传关联
表型编码规则:遗传共性维度与非遗传特异维度的解耦
通过UBHDD分离酵母与人类脑表型的遗传和非遗传子空间后,研究发现:遗传子空间的潜在维度(解释85%方差的主成分数)随性状采样迅速饱和(酵母约10维,人脑约15维),而非遗传子空间维度持续增长(图6a-b),印证“有限遗传维度+无限非遗传维度”理论。遗传维度被多性状重复利用(图6c-d),性状间遗传成分相关性显著高于非遗传成分(图6e-h),表明遗传因素通过多效性基因、共享通路、遗传连锁等机制驱动共性维度;而非遗传维度由环境异质性、发育可塑性、测量噪声等非遗传因素塑造,呈现高度性状特异性。该规则揭示表型空间的核心架构:复杂性状由少量遗传“基向量”的线性组合与大量环境“噪声向量”叠加形成,为表型组学提供了普适性理论框架。
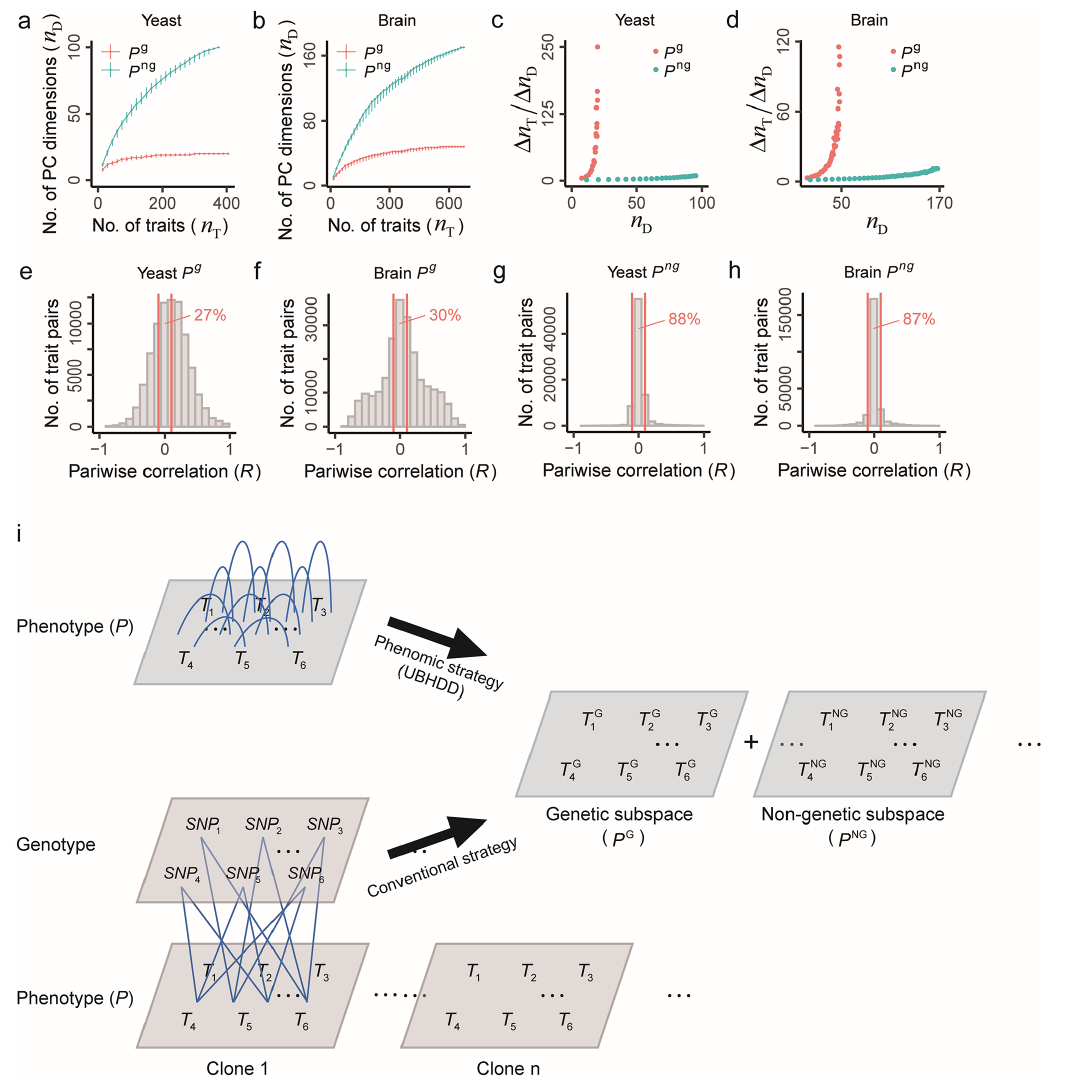
图6 遗传与非遗传子空间的维度差异
研究结论
本研究通过构建基于非相关性的高维依赖分解方法(UBHDD),首次揭示了表型空间中遗传(PG)与非遗传子空间(PNG)的编码规则:遗传因素主导的遗传子空间由有限且复用性强的遗传维度构成,而非遗传因素塑造的非遗传维度具有近乎无限的性状特异性维度。通过酵母与人类脑表型组数据的验证,UBHDD在无需基因型信息下成功分离遗传与非遗传子空间,不仅解析了遗传相关性(如人脑左右不对称主要源于非遗传因素)、挖掘了数百个脑区与精神疾病的新遗传关联,还为表型组学奠定了“遗传驱动共性维度-环境塑造特异维度”的核心理论框架。这一发现突破传统“基因型驱动表型”的还原论局限,为复杂性状的系统解析提供了全新的数学工具与理论基础。
Abstract
Genotype and phenotype are both the themes of modern biology. Despite the elegant protein coding rules recognized decades ago in genotype, little is known on how traits are coded in a phenotype space (P). Mathematically, P can be partitioned into a subspace determined by genetic factors (PG) and a subspace affected by non-genetic factors (PNG). Evolutionary theory predicts PG is composed of limited dimensions while PNG may have infinite dimensions, which suggests a dimension decomposition method, termed as uncorrelation-based high-dimensional dependence (UBHDD), to separate them. We applied UBHDD to a yeast phenotype space comprising ~400 traits in ~1000 individuals. The obtained tentative PG matches the actual genetic components of the yeast traits, explains the broad-sense heritability, and facilitates the mapping of quantitative trait loci, suggesting the tentative PG be the yeast genetic subspace. A limited number of latent dimensions in the PG were found to be recurrently used for coding the diverse yeast traits, while dimensions in the PNG tend to be trait specific and increase constantly with trait sampling. A similar separation success was achieved when applying UBHDD to the UK Biobank human brain phenotype space that comprises ~700 traits in ~26,000 individuals. The obtained PG helped elucidate the genetic versus non-genetic origins of the left-right asymmetry of human brain, and reveal several hundred novel genetic correlations between brain regions and dozens of mental traits/diseases. In sum, by developing a dimension decomposition method we show that phenotypic traits are coded by a limited number of genetically determined common dimensions and unlimited trait-specific dimensions shaped by non-genetic factors, a rule fundamental to the emerging field of phenomics.
作者简介
通讯作者
贺雄雷,中山大学生命科学学院教授,中山大学生命科学学院院长,教育部长江学者特聘教授(2012年),国务院学位委员会学科评议组成员。曾获国家杰出青年基金(2012年)和国家万人计划科技创新领军人才项目(2019年)资助。2007年从美国密歇根大学(University of Michigan-Ann Arbor)获博士学位,同年通过中山大学“百人计划”引进回国担任生命科学学院教授。研究领域为进化遗传学,对基因重复,性染色体剂量补偿和癌症演化等问题开展过系统性工作;目前尝试将物种进化树的思想带入个体发育,通过发展高效细胞谱系标记技术,构建合子到成体的发育细胞谱系树(zygote-to-adult developmental cell phylogeny),从谱系的维度解析个体发育、组织器官稳态维持和肿瘤发生发展等重要过程。作为通讯作者在包括Science, Nature Methods, Nature Genetics, Nature Communications,PNAS, National Science Review, MBE等主流学术期刊发表研究论文三十多篇,并受邀为Science撰写观点文章(Perspectives)。
第一作者
王建国,中山大学逸仙班生物科学、数学与应用数学双专业学士;生物信息学博士,导师贺雄雷教授。现为中山大学生命科学学院博士后,受国家自然科学基金青年基金、广东省基础与应用基础研究基金青年项目等资助。研究兴趣为利用数学、统计和深度学习等方法发展生物系统的理论模型,阐述复杂疾病的遗传基础、胚胎发育的时空稳态和基因型-表型空间的结构和演化规律。于Journal of Genetics and Genomics、Molecular Biology and Evolution、Phenomics、Nature methods等发表论文。
https://wap.sciencenet.cn/blog-3558836-1496411.html
上一篇:Phenomics | 中国科学院上海营养与健康研究所汪思佳团队合作开发基于深度学习的舌象表型自动化平台TonguExp
下一篇:Phenomics | 中南大学湘雅二医院黎志宏教授团队综述:肉瘤 3D 模型,开启精准医疗新维度
全部作者的其他最新博文
- • Phenomics | 中山大学肿瘤防治中心曾木圣院士团队发表综述:肿瘤诊疗新靶点,整合素α6靶向技术引领精准医疗新时代
- • Phenomics | 上海交通大学医学院张孝勇教授团队开发磁共振成像降噪新方法,有潜力改善脑小血管病的诊断效能
- • Phenomics | 复旦大学倪挺教授团队揭示基因内含子多聚腺苷酸化调控细胞衰老新机制
- • Phenomics | 加拿大阿尔伯塔大学和复旦大学联合开发基于文本、音频和视频的多模态抑郁症检测与评估方法
- • Phenomics | 翟振国/蒋太交/张鹏团队合作开发自适应多基因风险评估模型提升汉族人群静脉血栓栓塞症的风险预测能力
- • 祝贺 | 《表型组学(英文)》成功入选2025年“中国科技核心期刊”