博文
使用SAMtools软件进行序列可视化展示
|
SAMtools序列可视化操作
1)准备 BAM 文件
# 使用参考序列构建索引,index为索引前缀
bowtie2-build reference.fa index

# 使用bowtie2进行比对
bowtie2 -x index -1 reads_1.fq -2 reads_2.fq -p 32 -S example_pair.sam
# 使用samtools将sam文件转换为bam文件
samtools sort example_pair.sam > example_pair.bam
2)使用 SAMtools 对 BAM 文件进行排序和索引(这是可视化展示的前提)
# 排序 BAM 文件
samtools sort example_pair.bam -oexample_pair.sorted.bam
# 创建 BAM 索引
samtools index example_pair.sorted.bam
3)可视化
# 使用 Samtools tview查看 BAM 文件的比对情况
samtools tview example_pair.sorted.bam reference.fa
# 运行后会进入santools tview的交互界面:
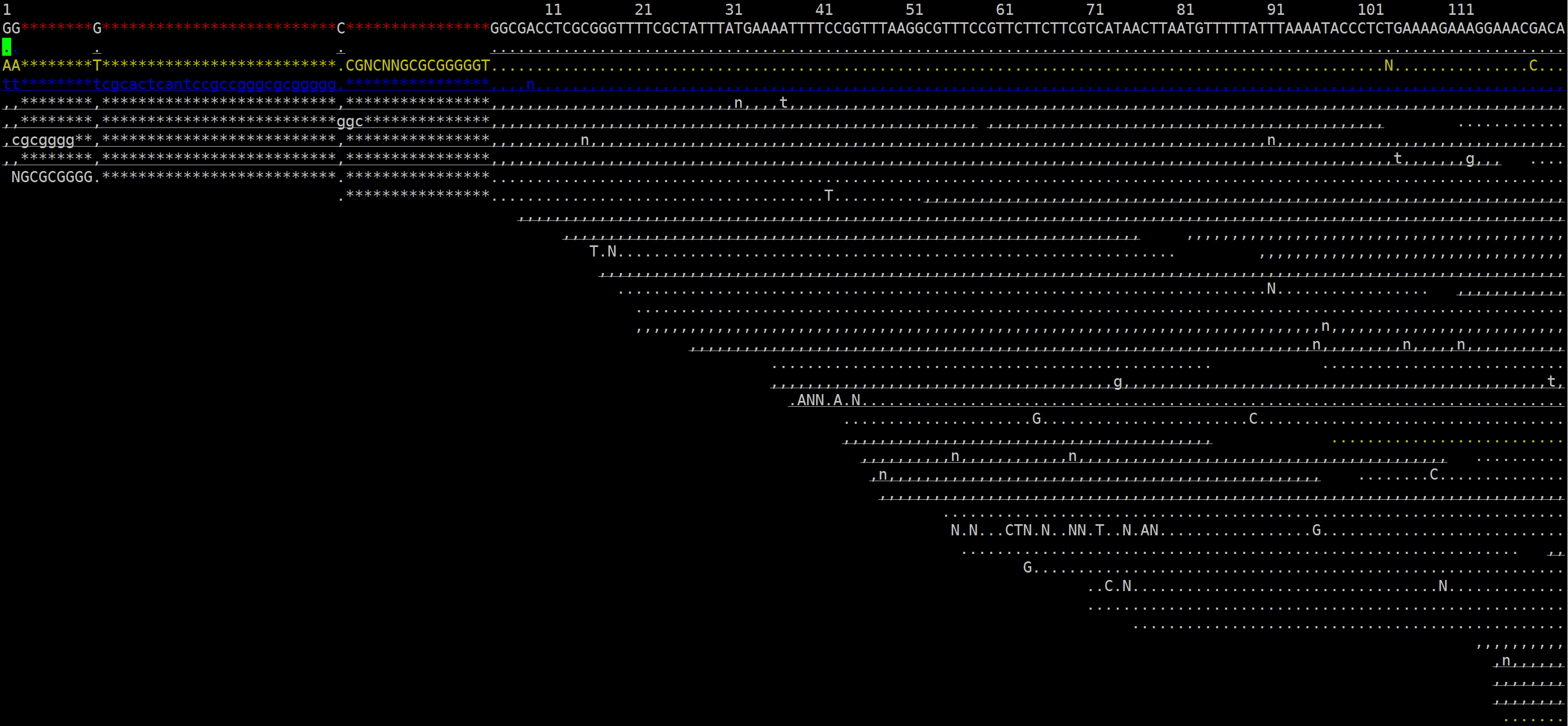
# 按下 “shift+?” 即可显示帮助菜单栏,如下图所示:
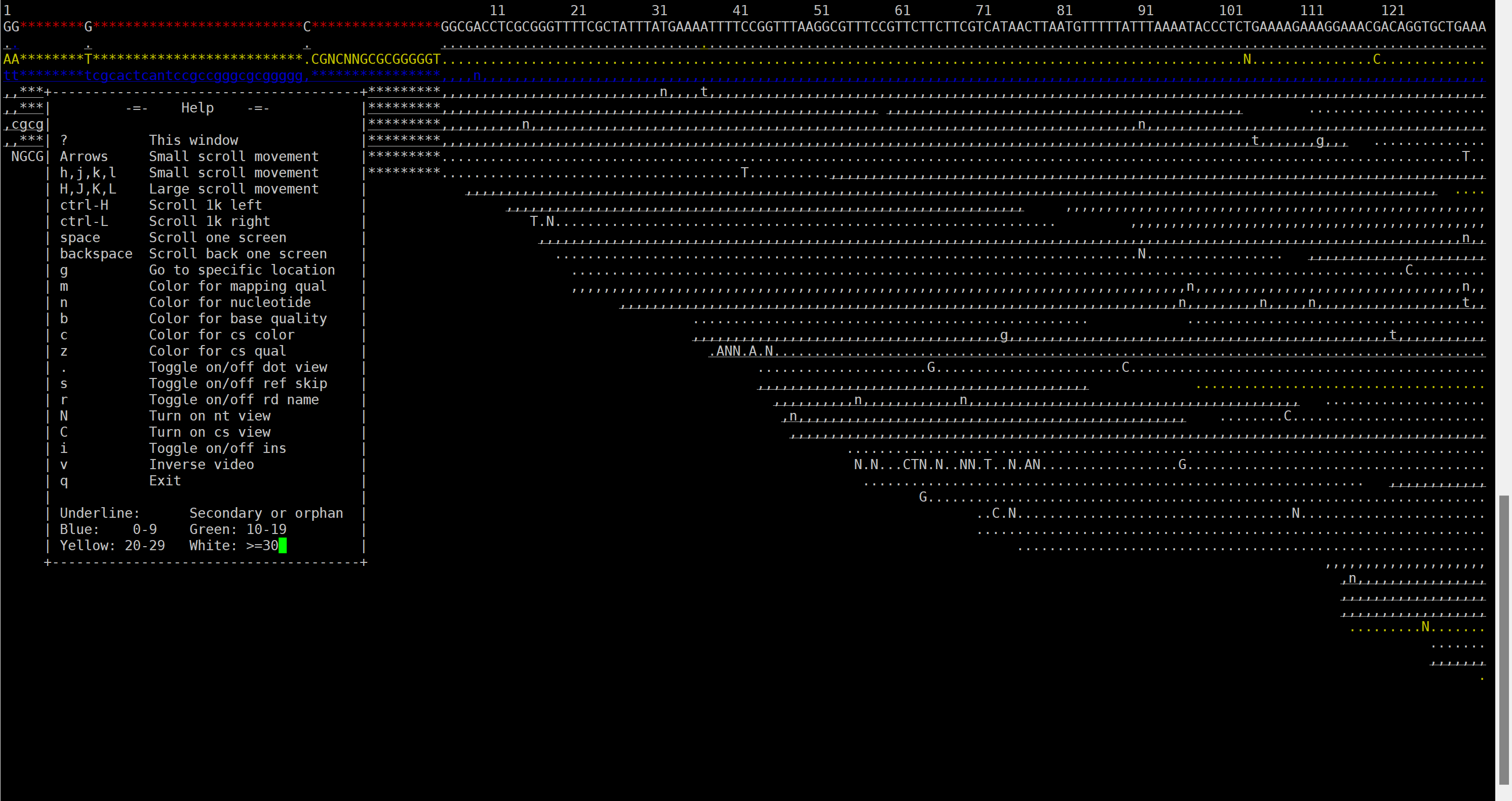
① 按下 g ,则提示输入要到达基因组的某一个位点
② 使用H(左)J(上)K(下)L(右)移动显示界面。
③ Ctrl+H 向左移动1kb碱基; Ctrl+L 向右移动1kb碱基
④ 可以用颜色标注比对质量,碱基质量,核苷酸等。
30~40的碱基质量或比对质量使用白色表示;
20~30黄色;
10~20绿色;
0~10蓝色。
⑤ 使用点号’.'切换显示碱基和点号;
⑥ 使用r切换显示read name
我们将持续分享微生物组学研究和生信分析相关的专业技能资料。推荐课程请搜索“密码子学院”。课程问题或个性化分析需求,请联系小唯(微信号:winnerbio01)。
https://wap.sciencenet.cn/blog-3447233-1495626.html
上一篇:借助Bowtie2软件分析基因组的测序深度
下一篇:Kaptive脂多糖血清型分类系统更新