博文
[转载]基于微生物组数据进行疾病预测的混合模型——BCGLMM
||

当前微生物组研究面临的主要挑战包括数据的成分特性、高维度、分类单元间的系统发育关系以及潜在的多种效应模式——即少数几个具有中等强度效应的微生物和大量具有微弱累积效应的微生物可能共同影响宿主健康。
为了应对这些挑战,近日,来自美国大通福克斯肿瘤中心和肯尼索州立大学等研究人员提出了一个新型模型——贝叶斯成分广义线性混合模型(Bayesian Compositional Generalized Linear Mixed Model, BCGLMM),其研究结果发表在《BMC Bioinformatics》上。
BCGLMM旨在提高利用微生物组数据进行疾病预测的准确性和解释性,它采用对数转换和对回归系数施加软性零和约束来处理成分数据;利用结合系统发育信息的结构化正则马蹄形先验来识别具有中等效应的关键分类单元;并通过引入基于样本间微生物组相似性的随机效应项来捕捉众多微小分类单元的累积影响。
模型在模拟数据和真实数据(炎症性肠病)的测试结果表明,BCGLMM在预测性能和识别相关微生物特征方面均优于现有的一些方法,证明了其在微生物组相关疾病预测领域的有效性和潜力。
方 法BCGLMM模型构建在广义线性混合模型(GLMM)的框架之上,专门针对微生物组数据的特性进行了调整。
数据预处理
1. 对OTU计数使用中心对数比(CLR)变换,回归系数施加软性零和约束。
2. 通过16S rRNA基因序列构建系统发育树,计算Bray-Curtis距离矩阵。
BCGLMM模型结构
BCGLMM模型主要由三个关键部分组成:
线性预测器(eta)
连接函数(g)
数据分布(p)
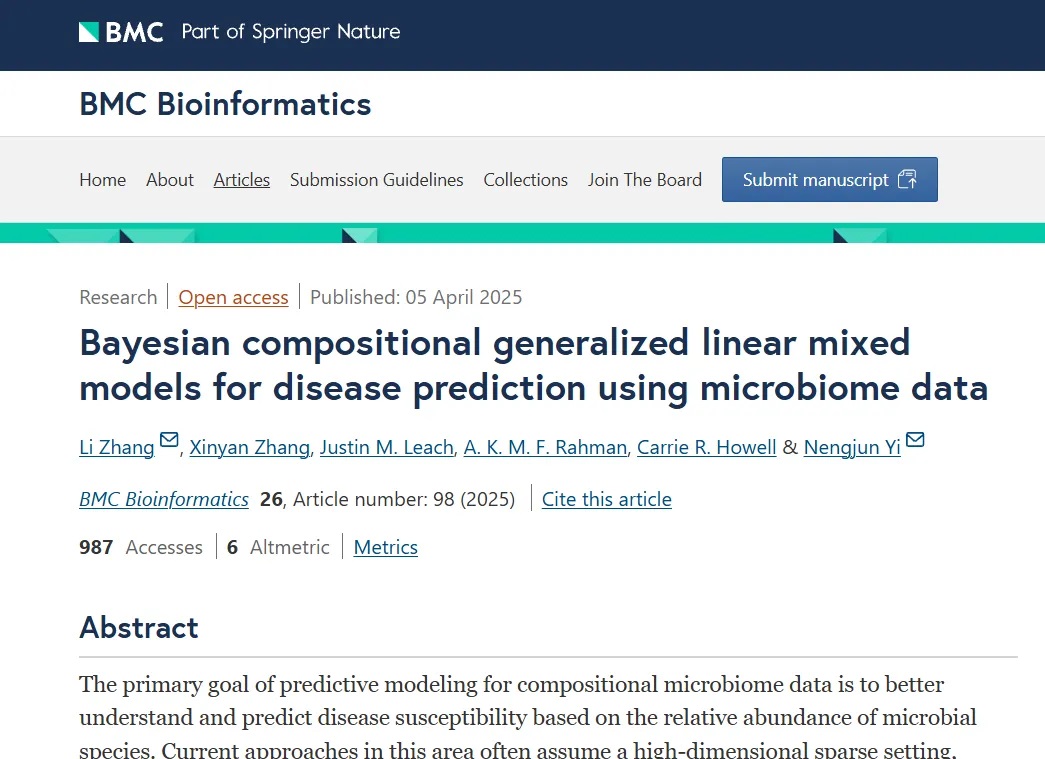
将线性预测器的输出通过sigmoid函数转为概率pi,然后根据预设的阈值转为二分类输出(如患病/健康)。
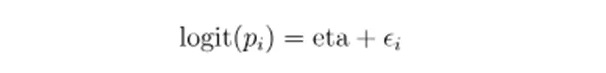
线性预测器(eta):
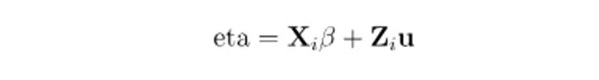

固定效应部分
结构化正则马蹄形先验(regularized horseshoe prior),适用于高维稀疏数据,先验分布约束参数空间。
整合系统发育信息,通过系统发育树构建分类群间的协方差矩阵,整合到先验协方差结构中。
随机效应部分
基于Bray-Curtis或UniFrac构建样本间距离矩阵。
通过高斯核函数将距离转换为相似性。
固定效应(如显著分类群)可能无法完全解释疾病风险,而大量微弱效应的分类群可能通过生态网络的整体扰动共同影响结果。随机效应用于建模这些样本层面的变异。两者结合后,模型既能抓住主要驱动因素,又能解释微生物生态系统的整体影响(如原文中IBD预测性能提升)。
后验分布输出
使用 R 包 brms(后端调用 rstan),通过 Hamiltonian Monte Carlo(HMC) 及其自适应变体 No-U-Turn Sampler (NUTS) 实现后验分布采样。
主 要 结 果1
模拟数据测试模型
固定样本数量为400,分类单元数目分别选择100、300、500。
固定中等效应数量,中等效应指的是一部分特定微生物特征与响应变量之间具有相对较强的、预先设定的关联。研究人员选择了0、6、12个特征。
固定小效应数量,小效应指的是大量微生物特征与响应变量之间可能存在的较弱的、广泛的关联。研究人员选择了占总特征数的20%、50%、70%。
组合不同样本数量、中等效应数量和小效应数量,分别生成连续和二元数据,共得到了27种场景,每个场景下,测试三种模型,分别为:
BCGLMM(考虑了样本相关的随机效应和预测变量相关性)
BCGLM(仅关注预测变量相关性)
BGLM(不考虑随机效应和预测变量相关性)
下表是研究人员提供的当中等效应数量为6时,每个模型在不同数据集和小效应比例下的性能评估结果:
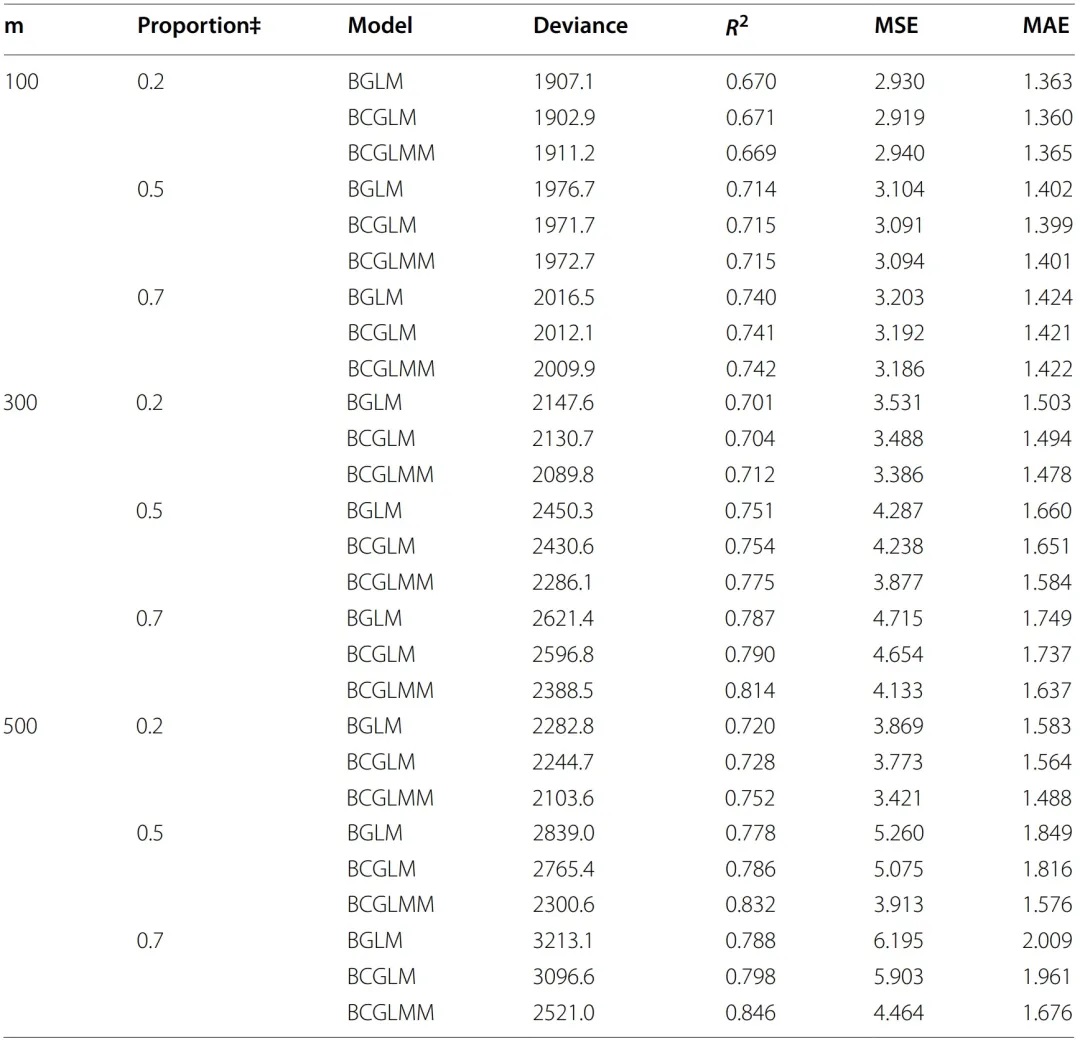
表1
结果显示,大部分场景下,模型性能从优到劣排序为:BCGLMM > BCGLM > BGLM
BCGLMM显示为R2最高、MSE和MAE最低。
当m=100时,小效应比例为0.2和0.5时,BCGLMM相比BCGLM和BGLM没有显示出明显优势,但当小效应比例达到0.7时,BCGLMM开始显示更好的性能;
当m介于300~500时,BCGLMM持续优于其他两种方法,且随着小效应比例的增加,这种优势趋势变得更加明显。
这些都说明BCGLMM中的随机效应能够捕获所有标记的综合小效应。
另外,研究人员给出的相同场景下的二分类结果,显示BCGLMM准确性更好(更高的AUC值),如下表:
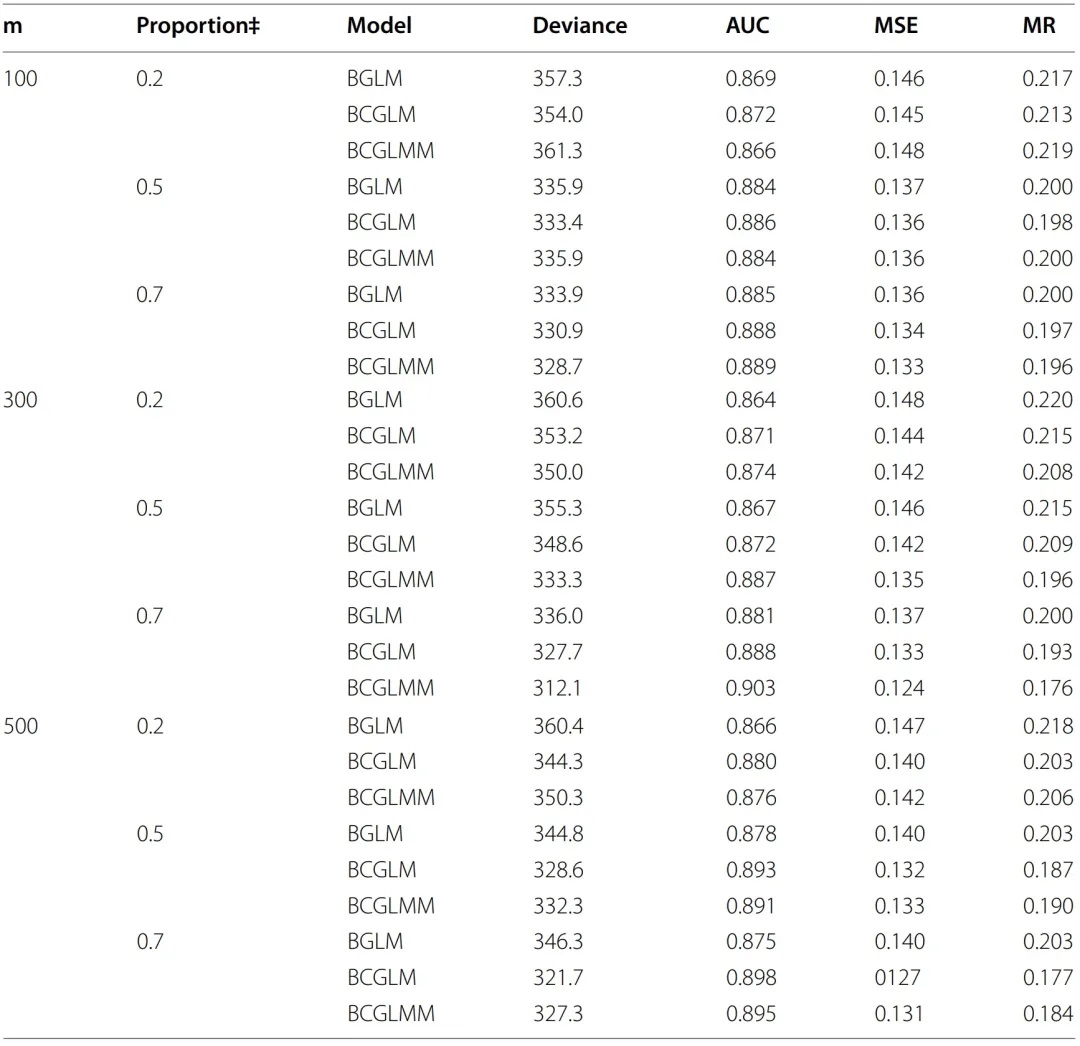
表2
对于其它中等效应数量的组合场景,结果如下图:
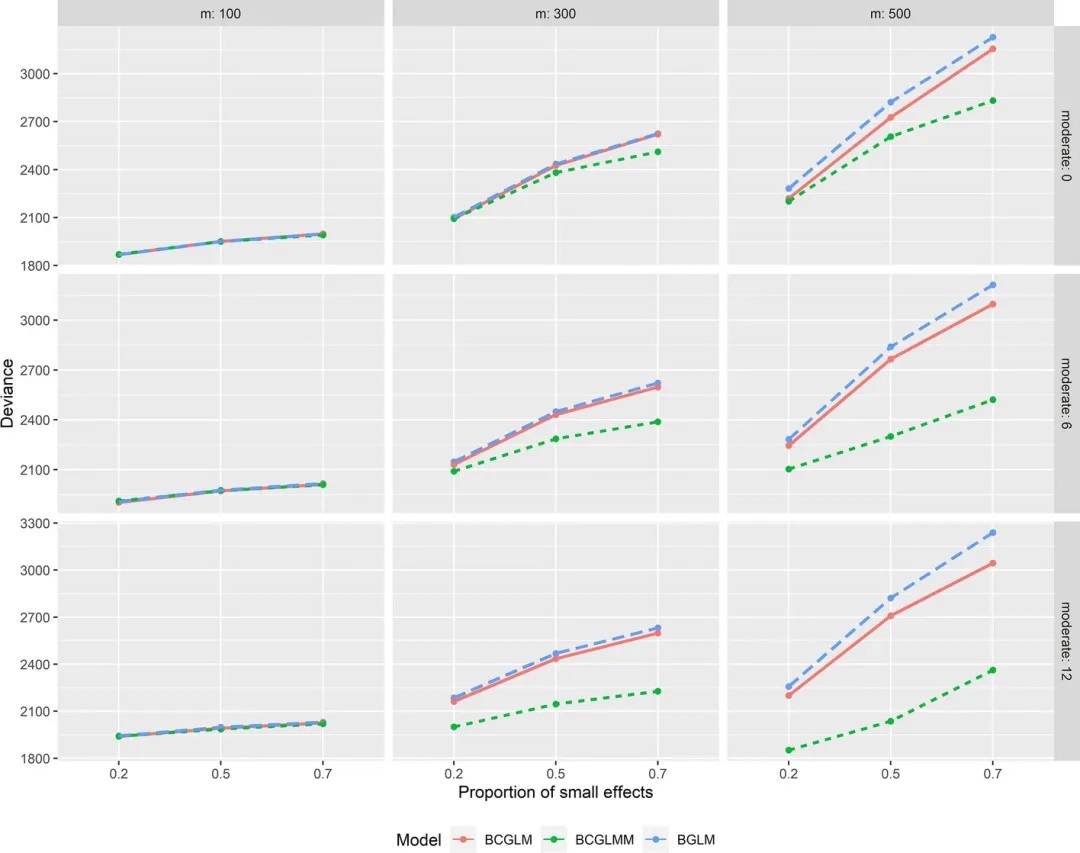
图1
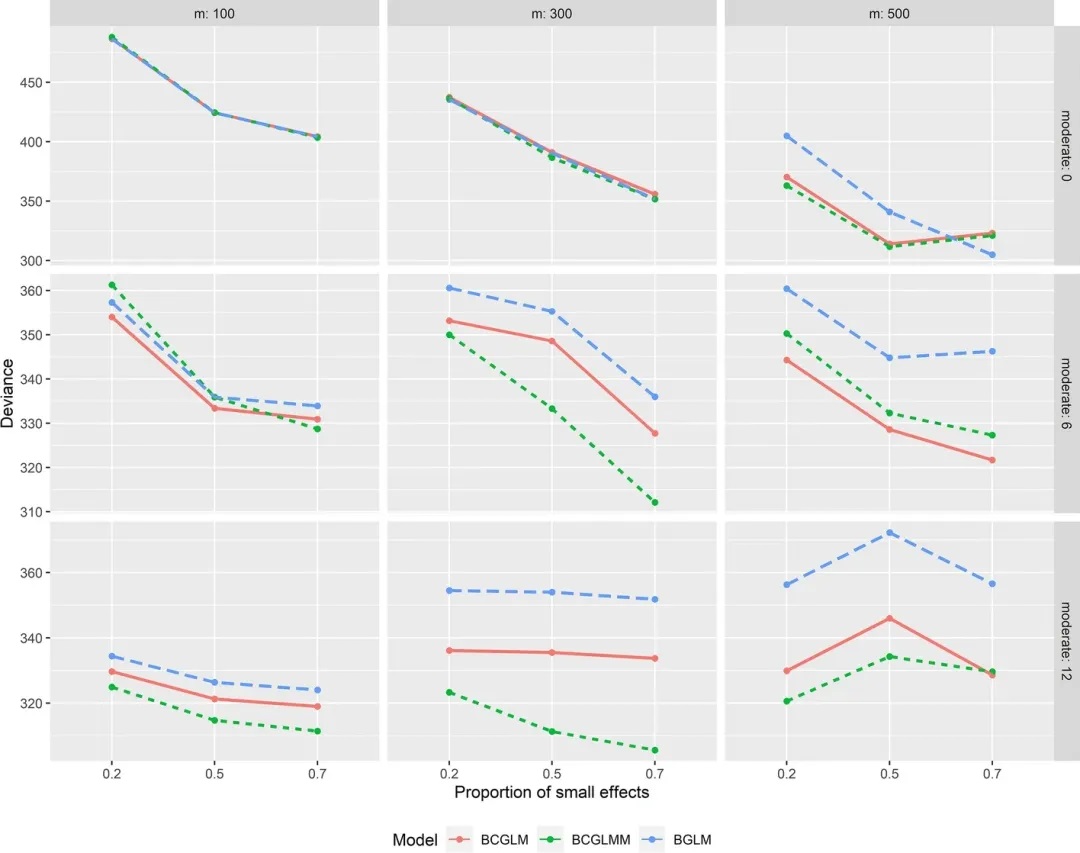
图2
图一对应表一(基于连续性结果的模型性能评估)
图二对应表二(基于二元结果的模型性能评估)
结果显示,在m介于300~500时,对比中等效应数量6,中等效应数量12场景下的BCGLMM模型性能更优。
2
真实数据测试模型
数据来自美国肠道项目(American Gut Project,AGP)的公开数据,旨在区分IBD患者和健康对照。含4684 个粪便样本和 1002 个物种数据。
基于“方法”对数据进行预处理后,分别应用BCGLMM(考虑了样本相关的随机效应和预测变量相关性)、BCGLM(仅关注预测变量相关性)、BGLM(不考虑随机效应和预测变量相关性)模型,比较三种模型的准确性。
根据AUC值判断,BCGLMM 、 BCGLM 和 BGLM 的交叉验证 AUC 值分别为 0.702 、 0.687 和 0.672,可以认为BCGLMM在真实数据集上的表现最优(AUC=0.702)。
结 论基于模拟数据和真实数据的测试结果,可以得出结论,同时考虑样本相关的随机效应和预测变量相关性的BCGLMM模型能够识别出与疾病相关的潜在微生物特征,二分类任务下,提升疾病预测的准确性。
BCGLMM模型的局限性在于计算成本较高,在大规模研究中可能面临内存和CPU时间挑战。综合看,推荐使用场景为:高维微生物组数据(特征数≥300),预期存在大量小效应的疾病预测任务,需要考虑系统发育关系的研究,对预测精度要求较高且能承受计算成本的项目。
该研究中的BCGLMM模型,突破传统稀疏模型的限制,首次系统性地解决了微生物组数据分析中的核心挑战,通过混合模型框架,能够识别中等效应并累积大量小效应,显著提升了疾病预测的准确性。
BCGLMM模型具有很大的转化潜力。
首先,该方法可扩展至多种疾病的预测和诊断,其高维数据处理能力为复杂疾病(如代谢综合征、自身免疫病)的微生物标志物挖掘提供了新的工具。
其次,结合样本随机效应的设计有助于解析宿主-微生物互作的个体异质性,可能推动个性化医疗中基于微生物组的风险分层策略。
随着单菌功能解析与宏基因组数据的深度融合,模型可进一步扩展至多组学整合分析(宏基因组、代谢组、转录组等),例如纳入菌株水平变异或代谢通路信息以提升预测精度。
该方法还可应用于不同样本部位的微生物组分析,如口腔、皮肤、呼吸道等,扩大临床应用范围。
总体而言,BCGLMM为微生物组驱动的精准医学提供了方法论基础,其兼顾统计学严谨性与生物学机制的框架有望成为探索微生物生态与疾病关联的核心技术之一。
主要参考文献
Zhang, L., Zhang, X., Leach, J.M. et al. Bayesian compositional generalized linear mixed models for disease prediction using microbiome data. BMC Bioinformatics 26, 98 (2025).
本文转自:谷禾健康
https://wap.sciencenet.cn/blog-2040048-1493422.html
上一篇:[转载]肠道神经免疫轴:神经元、免疫细胞和微生物之间的串扰
下一篇:[转载]从“菌”入手,助力睡眠:灵芝、茯苓、酸枣仁等的助眠之道