
氧级联反应:理解低氧适应的工具
摘要
低氧对全球约1%的人口构成生命威胁,同时也是导致多种心肺疾病、血液疾病和循环系统疾病患者发病率与死亡率升高的重要因素。然而,在相当一部分病例中,低氧适应以失败告终——因为潜在的适应通路往往与健康需求相冲突,进而引发疾病;在世界部分高海拔地区,这类疾病仍困扰着多达三分之一的常住人群。为助力理解适应与不适应的机制,本综述分析了从大气到线粒体的氧级联反应各环节,区分了生理性低氧(如高海拔导致)与病理性低氧(如基础疾病导致)相关的反应模式。研究旨在通过多学科视角评估人类对低氧的适应能力,将基因、分子和细胞功能与生理及病理结局相关联。结论认为:多数情况下,并非低氧本身引发疾病,而是机体对低氧环境的适应尝试;这印证了一种范式转变——当低氧适应过度时,便会转化为“不适应”。
参考文献
Samaja M, Ottolenghi S. The Oxygen Cascade from Atmosphere to Mitochondria as a Tool to Understand the (Mal)adaptation to Hypoxia. Int J Mol Sci. 2023 Feb 12;24(4):3670.
1. 引言
全球约1%的人口长期居住在海拔2500米以上地区,且主要分布在非西方国家[1]。这些人群持续面临的核心健康问题,源于呼吸氧气含量匮乏的大气。平均而言,高海拔常住人群中多达10%会患上与氧气不足(即低氧)相关的疾病[2]。即使在海平面,低氧也普遍存在,它导致大量急性呼吸系统疾病(ARDS)、慢性肺部疾病(COPD)、肺气肿、肺部肿瘤、贫血、心力衰竭及其他数百种“氧供需失衡”相关疾病患者的发病率和死亡率升高。这类疾病给人类带来难以承受的痛苦、生命损失和经济负担——仅2010年一年,美国因COPD产生的负担就高达499亿美元[3]。在所有影响人类健康的因素中,低氧的独特之处在于它兼具“生理性”(如高海拔所致)和“病理性”(如基础疾病所致)双重属性。生理性低氧可能诱导机体产生某种形式的适应,而对环境应激因素的适应能力,正是达尔文物种进化理论中的核心特征。然而,鉴于生理性和病理性低氧相关临床表现的高发率,人类能否真正适应低氧,目前仍不明确。
本综述的核心目标是聚焦机体对“氧气不足”这一挑战的适应能力。为此,我们将从基因层面到呼吸、循环系统功能层面,分析低氧应答的潜在机制,重点关注与适应最相关的环节。为实现这一目标,我们将重新审视“氧级联反应”,将其作为理解生理性和病理性低氧发生机制及后果的工具。文中将聚焦影响低氧适应的通路,暂不讨论单纯的低氧应答相关内容;在数据选择上,将优先采用人类相关研究结果,而非动物实验或体外实验数据。在描述低氧程度时,将同时标注“海拔(米)”及对应的“有效氧浓度(%)”(海平面氧浓度定义为21.0%)。
2. 氧气与低氧
氧气的发现归功于18世纪的三位科学家。瑞典-德国药物化学家卡尔·威廉·舍勒(Carl Wilhelm Scheele,1742–1786)是首位发现氧气的人,但因其学术影响力较低且缺乏国际竞争力,错失了“氧气发现者”的优先权[4]。英国博学家约瑟夫·普里斯特利(Joseph Priestley,1733–1804)兼具化学家、自然哲学家、分离派神学家、语法学家、多学科教育家和自由政治理论家等多重身份,他将科学研究与当时关于“燃素”的争论相结合,却因与英国国教的冲突被迫移居美国。尽管其研究成果的发表时间晚于舍勒,但他仍被认定为“氧气发现者”并获得优先权[5]。最后,法国贵族兼化学家安托万·拉瓦锡(Antoine Lavoisier,1743–1794)与玛丽-安妮·拉瓦锡(Marie-Anne Lavoisier)共同创造了“氧气”(oxygen)这一术语——该词源于“产生酸性的物质”这一当时被误解的概念(希腊语中,ὀξύς(oxys)意为“酸性”,-γενής(-genēs)意为“产生者”)。最终,安托万在法国大革命期间被送上断头台处决[6]。此外,17世纪的两位科学家——英国的约翰·梅豪(John Mayhow,1661–1679)和波兰的米哈乌·森迪沃吉乌斯(Michael Sendivogius,1566–1636)的贡献也值得提及,尽管他们的研究成果可能不足以使他们获得“氧气发现者”的头衔[7]。
2.1 高海拔环境中的氧气
根据道尔顿定律,氧气分压(PO₂)可通过以下公式表示为“氧浓度(%O₂)、大气压(BP)和水蒸气压(PH₂O)”的函数:
PO₂ = (BP − PH₂O) ÷ 100 × %O₂ (1)
大气压和海拔是决定大气PO₂的主要因素(图1),因为水蒸气压仅取决于温度(37°C时为47 mmHg),且除南极洲(物理因素导致氧浓度略有下降)外,地球任一纬度的氧浓度均保持稳定(20.9460%)。在海拔3233米/有效氧浓度14.3%的 Dome C 康科迪亚科考站,空气中的氧浓度略降至20.82–20.90%[8],这使得其“有效氧浓度”相当于海拔3800米/13.2%的环境[9]。
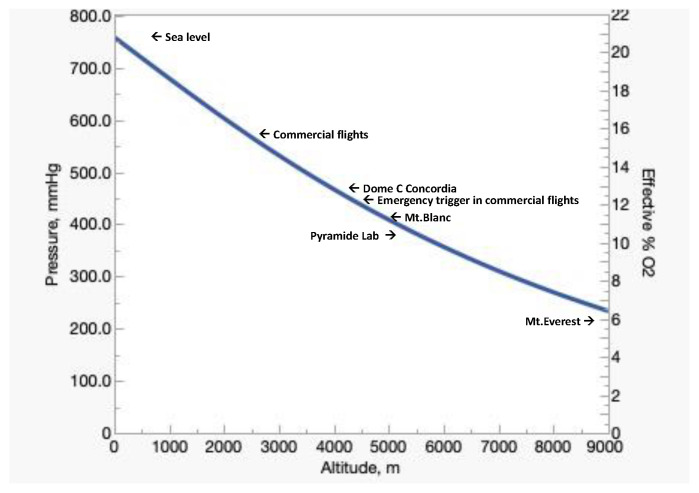
图1. 地球大气中,大气压(左Y轴)与有效氧浓度(右Y轴)随海拔变化的规律
数据来源于国际标准大气(ISA)模型(该模型未计入水分)。
低氧环境会显著影响运动能力。图2展示了“静息状态”和“最大摄氧量(VO₂max)”随海拔的变化:静息摄氧量不受海拔影响,始终维持在约3.5 mL O₂/kg/min(即1个代谢当量,MET);与之相反,最大摄氧量则从海平面“训练水平极高者”的约18 MET,逐渐下降至珠穆朗玛峰(海拔8848米/氧浓度6.6%)峰顶附近的3–4 MET——这一水平仅能支持步行、机床操作、站立作业或高尔夫等轻度活动。因此,高海拔环境中氧气可用性的降低,使得人体仅能完成极轻度任务,更不用说在该环境下攀登高峰及抵御极端低温了。此外,静息摄氧量占最大摄氧量的比例,从海平面的5%升至海拔8848米/氧浓度6.6%环境下的约30%——这意味着在极端高海拔环境中,吸入氧气的很大一部分仅能满足机体基础代谢需求。
目前,珠穆朗玛峰峰顶(海拔8848米)的实测大气压为253 mmHg[10],这一压力或许足以维持基础代谢。然而,平流层气球测量数据显示,大气压会因纬度和季节不同而存在显著差异[11]。
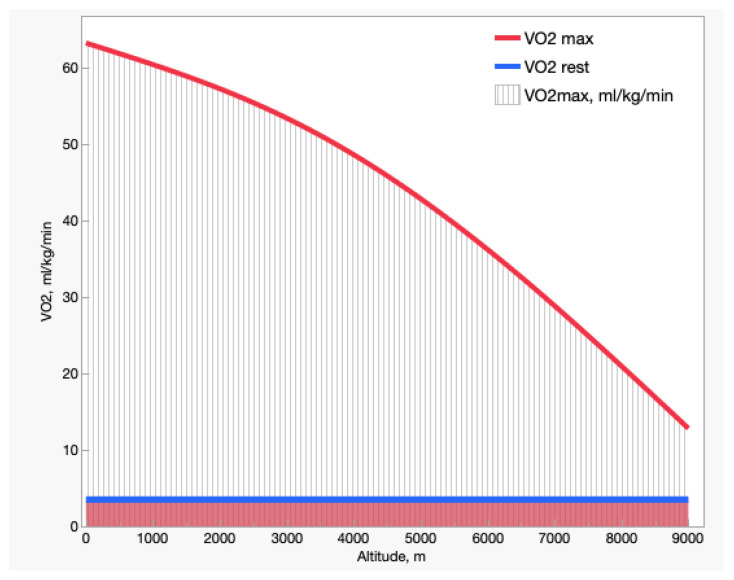
图2. 最大摄氧量(红色)与静息摄氧量(蓝色)随海拔变化的规律
数据改编自文献[12],该文献汇总了“海拔0–9000米范围内(含真实海拔与模拟海拔)、不区分训练水平”的146项研究的平均值。虚线区域代表可用于运动的氧气量。
2.2 低氧的定义
低氧被定义为“氧气供应与氧气需求的失衡”。这一定义适用于从“完整生物体”到“细胞”的所有组织层次,且涵盖生理和病理两种场景。尽管高海拔低氧的基本概念如今已被用于解释多种人类疾病,但直到近年,“低氧”仍几乎仅被视为“高海拔环境、载人热气球飞行,或山区临时/永久居住者”面临的问题(表1)。
表 1. 低氧对人类影响的部分历史记载
时间 | 事件描述 |
公元前 30 年 | 张骞出使西域途经兴都库什山脉时,首次记载低氧效应:“…… 有大头痛山、小头痛山…… 人至则体热、面色苍白、头痛呕吐……”[13] |
1590 年 | 西班牙耶稣会士 Padre Jose de Acosta 穿越安第斯山脉时记录:“许多士兵在穿越帕里亚卡卡山口(海拔 4800 米 / 氧浓度 11.6%)时患病…… 我被一种奇特的剧痛击倒在地,才意识到这里空气稀薄,不适合人类呼吸……” |
1862 年 | 亨利・考克斯韦尔(Henry Coxwell)与詹姆斯・格莱舍(James Glaisher)在伍尔弗汉普顿上空升至海拔 8850 米时记录:“…… 四肢麻痹、失明、言语困难、失去意识……”—— 返回地面后,两人完全康复。 |
1875 年 | 西维尔(Sivel)、蒂桑迪耶(Tissandier)与克罗切 - 斯皮内利(Croce-Spinelli)乘热气球升至海拔 8500 米:三人均失去意识,仅蒂桑迪耶生还,但听力永久受损。 |
1897 年 | 安杰洛・莫索(Angelo Mosso,1846–1910)出版《阿尔卑斯山人类生理学》(Fisiologia dell’Uomo sulle Alpi,由 Fratelli Treves 出版社出版),书中描述了在海拔 4554 米 / 氧浓度 12.0% 的玛格丽塔皇后小屋(Capanna Regina Margherita)开展的实验,涉及肌肉力量、呼吸、血液循环、心肌功能、营养、肺活量、高山病及神经生理问题。 |
1916 年 | 亚历山大・凯拉斯(Alexander Kellas,1868–1921)在《皇家地理学会会刊》发表文章,分析攀登喜马拉雅山脉最高峰的可能性;在其生前未发表的手稿中,他提出 “身体天赋出众且训练充分的人,无需吸氧即可攀登珠峰”[14]。 |
1953 年 | 埃德蒙・希拉里(Edmund Hillary)与丹增・诺尔盖(Tensing Norkay)借助补充氧气,首次登顶珠穆朗玛峰(海拔 8848 米 / 氧浓度 6.6%)。 |
1978 年 | 莱因霍尔德・梅斯纳(Reinhold Messner)与彼得・哈贝勒(Peter Habeler)在不借助补充氧气的情况下,首次登顶珠穆朗玛峰。 |
1979–2022 年 | 珠穆朗玛峰(海拔 8848 米 / 氧浓度 6.6%):累计约 270 名登山者死亡,多数死因直接或间接与低氧相关。 |
1987 年 | 罗伯特・M・温斯洛(Robert M. Winslow,1941–2009)与卡洛斯・蒙赫・卡辛内利(Carlos Monge Cassinelli,1921–2006)出版《低氧、红细胞增多症与慢性高原病》(约翰・霍普金斯大学出版社,ISBN 0-8018-3448-1),书中阐述了全球不同高海拔地区原住民慢性高原病的发病机制。 |
低氧的严重程度可根据“动脉PO₂”和“暴露时间”分为轻度至重度[15],但触发低氧状态的“PO₂阈值”尚未明确——这是因为该阈值受多种病理生理因素影响(如细胞代谢率、摄氧量、心输出量、氧提取率等),导致个体差异极大。例如,部分COPD、心力衰竭和贫血患者在海平面(0米/21.0% O₂)环境下即存在低氧;而未经训练的健康人可在海拔3300米/14% O₂环境下短期停留,且无明显不适;训练水平极高者甚至能在海拔7500米/8.0% O₂环境下存活数小时至数天。在细胞层面,当前通用实验方法[16]建议将培养细胞暴露于“相当于海拔20000米/1.0% O₂”的环境中,以模拟低氧条件。
即使在同一生物体内,不同器官的“摄氧量(VO₂)、毛细血管血流量和氧提取率”也存在显著差异。这意味着,针对相同程度的低氧,不同器官的敏感性会因其生理状态不同而有所区别。例如,在相同低氧条件下,大鼠不同器官在“氧感知机制激活”和“凋亡启动”方面的应答存在差异[17]。因此,可推断:在任一低氧程度下,脑、心脏、运动中的肌肉等高代谢率器官,会比“氧消耗与灌注更匹配”的低代谢率器官更早出现低氧。
2.3 低氧的临床表现
如何判断生物体是否已适应某种挑战?原则上,若受挑战群体“未出现该挑战相关疾病、可正常存活并繁殖”,则可认为其已适应。当低氧构成挑战时,相关疾病可分为急性和慢性两类,分别对应急性高原病(AMS)和慢性高原病(CMS)这两种复杂病理状态。全球高海拔常住人群中,CMS的发病率高达10%[2,18],且存在显著地域差异。
CMS由卡洛斯·蒙赫·梅德拉诺(Carlos Monge Medrano)首次描述[19],目前被视为评估“慢性低氧适应”的重要标志。作为一种由“持续性氧气不足”引发的复杂疾病,其主要致病机制是“过度红细胞生成(即红细胞增多症)”——由此产生的一系列后果统称为CMS[20]。因此,通过监测血液学指标变化,可评估低氧适应情况[21,22]。其他有效的“不适应标志物”包括高海拔地区常见的肺动脉高压(PH)(常伴随右心室肥厚和衰竭);此外,CMS相关的神经心理功能障碍也值得关注,因其可能影响儿童的学业表现[23]。为部分抵消这类影响,有研究提出“氧气调节”方案:利用合成沸石吸附空气中的氮气以提高氧浓度——研究发现,氧浓度每增加1%,相当于海拔降低300米[24]。
3. 氧级联反应
尽管存在重要局限性[25],“氧级联反应”仍是描述“氧气从大气到最终使用者——线粒体”转运路径的有效模型(图3)。图中蓝色曲线代表“常氧健康状态下”,PO₂沿转运路径逐步下降的过程:相邻组织间氧梯度的建立,确保了氧气沿级联路径的正常流动,且每个梯度的宽度必须足以保证线粒体获得充足氧气[26]。红色曲线代表“大气PO₂降低1/3”(相当于海拔3400米/13.9% O₂环境)时的情况:这种PO₂下降会导致“各级梯度宽度减小”,进而减少线粒体的氧气供应——因此,所有组织界面必须协同作用,才能确保氧气的正常输送。
此外,即使环境PO₂正常,若“氧级联路径中某一界面出现氧通量阻力位点”,也会导致线粒体氧气供应减少。这种情况表现为“相邻组织间PO₂梯度变宽”,且阻力位点通常与“将常氧环境转化为下游低氧环境”的疾病相关。由此引发的“病理性低氧”与“生理性低氧”的区别在于:前者常伴随炎症、氧化还原失衡等附加因素。
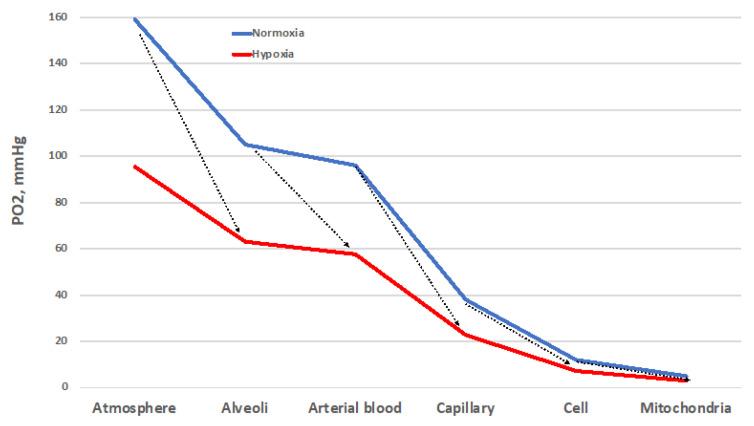
图3.常氧与低氧状态下的氧级联反应
常氧状态(蓝色)与低氧状态(红色,代表海拔3400米/氧气浓度13.9%的环境)下的氧级联反应。箭头指示氧通量的阻力位点,这些位点会增大血氧分压(PO₂)梯度,可能将常氧环境转变为低氧环境。其他解释详见正文。
物种进化的一个关键特征是对环境变化的适应能力。近年来,心脏、神经和肺组织对低氧环境的初始适应(即习服)机制已逐渐明确[27],但判断长期适应是否启动的标准应包括以下特征:出现高度适应环境的复杂性状(这种适应性不太可能随机产生),且这些性状能帮助个体存活和繁殖。
“适应生理学”的概念在很大程度上受限于该术语语义相关的问题[28]。尽管普遍认为“适应”涉及对环境挑战建立代偿性应答,但并非所有对挑战的应答都具有适应性。因此,根据“增益”(gain)大小,生物对挑战的应答可分为三个水平:不足(增益<100%)、理想(增益=100%)和过度(增益>100%)(图4)。本文将分析图3所示各阻力位点对低氧挑战的应答,以探讨“人类能否真正适应低氧”这一核心问题。
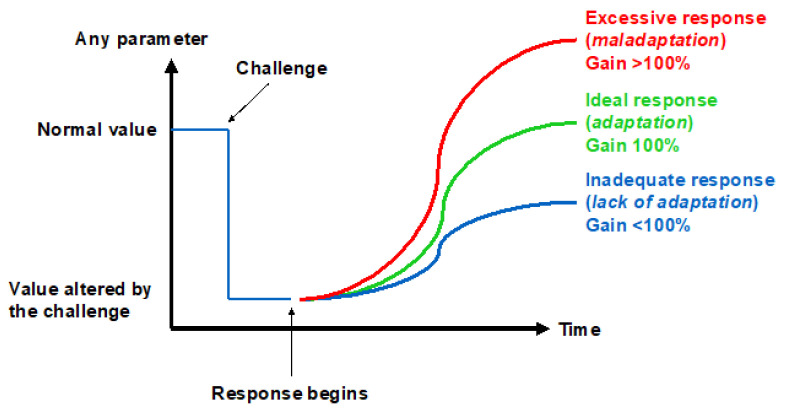
图4. 应对挑战的三种方式
3.1 细胞-线粒体界面
3.1.1 低氧诱导因子
威廉·凯林(William Kaelin)、彼得·拉特克利夫(Peter Ratcliffe)和格雷格·塞门扎(Gregg Semenza)因揭示细胞感知和适应氧气可用性的机制,共同获得2019年诺贝尔生理学或医学奖,该机制也得到全球学界的广泛认可。这种普遍存在的机制构成了“氧感知功能”,可调控细胞对缺氧的大部分(而非全部)应答。
低氧诱导因子(HIF)是转录因子家族,也是氧感知系统的核心组成部分。它通过诱导或抑制数百个基因的表达发挥作用,这些基因编码的蛋白质参与多种生理过程,包括代谢(如糖酵解)、形态发生(如血管生成)、细胞增殖与存活(如凋亡、细胞周期调控)及分子合成(如促红细胞生成素生成)等。
HIF的活性仅有一小部分受基因表达调控,其主要调控依赖脯氨酰羟化酶(PHD)和HIF抑制因子(FIH):在氧气存在时,PHD可使HIF的两个脯氨酸残基羟化,FIH则可使HIF的天冬酰胺残基羟化[29]。羟化后的HIF会发生泛素化,从而失去调控基因表达的活性。
HIF的β亚基为组成型表达,不响应氧气变化;而对氧气敏感的α亚基已发现至少三种亚型。其中研究最广泛的HIF-1α和HIF-2α具有48%的序列同源性,但在不同组织中的表达存在差异:例如,HIF-2α(又称内皮PAS结构域蛋白1,EPAS-1)主要在血管内皮细胞[30]、脑、心脏、肺、肾、肝、胰腺和肠道组织中表达[31]。尽管HIF-2α调控的基因数量少于HIF-1α,但它在红细胞生成中发挥重要作用[32,33]。此外,HIF-1α更易在短期强低氧环境中高表达,而HIF-2α则在长期轻度低氧环境中活性更高[34]。
HIF活性随氧气可用性的调控,依赖于PHD和FIH对PO₂变化的敏感性——这种敏感性通过米氏常数(KM)体现,即反应速率达到最大速率一半时的PO₂。细胞内平均PO₂范围为7-20 mmHg(1.0%-2.5% O₂),在此范围内,FIH(KM=50-80 mmHg/6.5%-10.5% O₂)作为生理性氧传感器的动态效能优于PHD(KM=120-210 mmHg/15.7%-27.6% O₂)[29,35,36]。但需注意,KM值受检测所需底物肽链长度的显著影响,因此解读时需谨慎。此外,要全面理解HIF在低氧适应中的作用,还需评估“HIF活性-PO₂”曲线的斜率,但目前尚无相关关键数据。
要明确HIF的氧感知机制规律,并将其视为低氧适应的关键因子,不仅需要获取PHD和FIH氧亲和力的动力学数据,还需研究HIF在长期及慢性低氧环境中的稳定性。体外实验(多在培养的肿瘤细胞中进行,因长期低氧问题在肿瘤生长中尤为突出)可部分解答这一问题,但现有数据存在显著矛盾:在神经母细胞瘤细胞中,缺氧超过72小时后,HIF-1α和HIF-2α蛋白均会失稳[37];有研究提出,在肿瘤发展过程中,急性低氧首先稳定HIF-1α,约24小时后HIF-1α水平下降,由HIF-2α替代,随后HIF-2α缓慢减少,再由其他低氧相关因子替代[38]。
一项针对人类微血管内皮细胞的研究跟踪了14天低氧暴露过程,发现HIF-2α相关的信使RNA(mRNA)表达持续时间长于HIF-1α,但缺乏更能反映HIF在低氧适应中作用的蛋白质水平数据[39]。急性与慢性低氧可引发完全不同的低氧相关应答:例如,急性低氧影响196个基因的表达,而慢性低氧导致4149个转录本变化,两者共有的差异基因仅144个[40]。转录组的这种显著变化(可能有助于阐明HIF在低氧适应中的作用),可能源于不同HIF亚型激活的下游转录效应差异[41]。事实上,对48小时低氧环境中HIF-3α(HIF的一种亚型)mRNA表达的研究发现:HIF-1α mRNA在4小时达到峰值,而HIF-3α mRNA则在HIF-1α水平下降时开始升高[42]。
体内肿瘤中PO₂的自发性波动,进一步增加了评估HIF在长期低氧中作用(及其在低氧适应中参与度)的复杂性;此外,低氧水平和暴露时间的实验方案差异极大(从30分钟到数周不等)[43],也导致结果难以统一。在“海拔5900米/氧气浓度10.0%、无复氧、持续14天”的慢性低氧环境中,健康大鼠的脑[44,45]、肌肉[46]和心脏[47]组织中HIF-1α蛋白水平升高,但心脏组织的HIF-1α mRNA水平未受影响[48]。
综上,由于HIF可能调控数百个基因的表达(约占人类基因组的2.6%[49]),目前仍难以判断这些活性是否均能转化为有效的低氧适应模式,因此“HIF是否参与适应过程”这一问题尚未得到明确解答。
3.1.2 抗氧化防御
氧气除了为氧化磷酸化供能外,还可通过非酶促(如线粒体解偶联)或酶促[50]过程生成高活性物质——活性氧(ROS)。ROS水平过高时,会因高反应活性产生毒性;但在生理水平下,ROS可调控多种通路,几乎参与所有细胞功能的控制。ROS与氧化应激的产生直接相关,而氧化应激是“活性物质生成”与“抗氧化防御”失衡的结果。
线粒体解偶联是低氧导致ROS生成的主要(但非唯一)途径[51]。功能异常的线粒体与质膜中的NADPH氧化酶之间存在“串扰”[52],使该酶成为额外的ROS来源[53]。其中,NADPH氧化酶4可直接响应缺氧[54],是低氧环境中ROS的主要来源[55,56]:在“海拔5900米/氧气浓度10.0%、持续4周”的低氧环境中,小鼠脑内NADPH氧化酶4的水平几乎翻倍[57]。
NADPH氧化酶4的活性可被红细胞生成素2相关因子2(Nrf2)拮抗:大鼠脑内的Nrf2可响应12小时低氧[58],并上调抗氧化防御机制[59]。另一种保护因子——蛋白激酶B(又称Akt),是一种丝氨酸/苏氨酸特异性蛋白激酶,可通过抑制凋亡发挥神经保护作用[60,61];即使在慢性低氧环境中,Akt也能体现保护功能,其作用甚至可能超过Nrf2引发的保护效应[57]。
在“海拔5900米/氧气浓度10.0%”的轻度低氧环境中,ROS生成和防御机制均会增强,但至少在4周低氧暴露期间,ROS生成的增幅更大,导致氧化还原失衡加剧,并出现明显的氧化应激迹象[57]。
谷胱甘肽是一种三肽硫醇,参与细胞对ROS的防御,可能也发挥重要作用。氧化型谷胱甘肽/还原型谷胱甘肽(GSSG/GSH)的比值可反映细胞的氧化还原状态[62];神经元对ROS的高敏感性,可能与脑内谷胱甘肽过氧化物酶活性相对较低有关[63]。在“海拔8500米/氧气浓度7.0%、持续6小时”的低氧环境中,大鼠脑提取物中的GSH水平和谷胱甘肽过氧化物酶活性均下降,表明低氧会显著损害谷胱甘肽构成的抗氧化防御[64]。
已知高海拔暴露会增强ROS生成,超过细胞防御能力,导致脂质、蛋白质和DNA损伤,进而引发或加重包括慢性高原病(CMS)在内的多种疾病[65]。对氧化挑战的适应需要较长时间,而体育锻炼通常会加剧这种挑战的严重程度[66]。例如,在“海拔3800米/氧气浓度13.2%”的环境中,人类体内氧化应激的间接标志物在低氧暴露第20天达到峰值,并在随后10个月内持续升高[67]。
神经元和大脑对ROS高度敏感[68,69],这是因为其氧耗量(VO₂)高而抗氧化防御能力弱[70]。因此,低氧诱导的氧化应激最显著的副作用之一,是频繁观察到的记忆和认知功能障碍——这一结果不仅在神经学研究中被证实[71],在动物模型中也得到验证[72]。
认知障碍可能与“抗氧化防御不足导致的氧化应激启动”直接相关:例如,在“海拔6100米/氧气浓度9.8%、持续7-14天”的低氧环境中,大鼠ROS水平升高,且与海马体、皮层和纹状体中的神经型一氧化氮合酶(nNOS)表达增加、神经退行性变及DNA片段化呈正相关[73];同样,“海拔5900米/氧气浓度10.0%、持续2周”的低氧会导致显著的神经元凋亡(这是明显脑损伤的标志)[44,45]。
综上,应对氧化还原挑战需关注两个方面:低氧诱导的氧化应激增强,以及内源性抗氧化防御的建立——后者可能无法充分发挥作用(尤其是在长期低氧环境中)。从低氧适应的角度来看,抗氧化防御所代表的潜在适应性应答,似乎难以达到预期目标。
3.1.3 生物能量学
人体吸入的氧气中,绝大多数(约97%)用于长链反应的最终步骤——氧化底物(主要是葡萄糖、脂肪酸,部分器官还包括酮体)(图5)。细胞内的氧气(通过简单扩散进入,收缩细胞中则通过与肌红蛋白(Mb)结合运输)最终被细胞色素氧化酶利用,生成三磷酸腺苷(ATP)形式的生物能量。
本文暂不考虑无氧乳酸通路的贡献(即磷酸肌酸穿梭和腺苷酸激酶反应产生的生物能量),因为这些通路仅能在有限时间内提供ATP。显然,线粒体形态和密度的任何变化,都不可避免地会导致生物能量途径的改变。
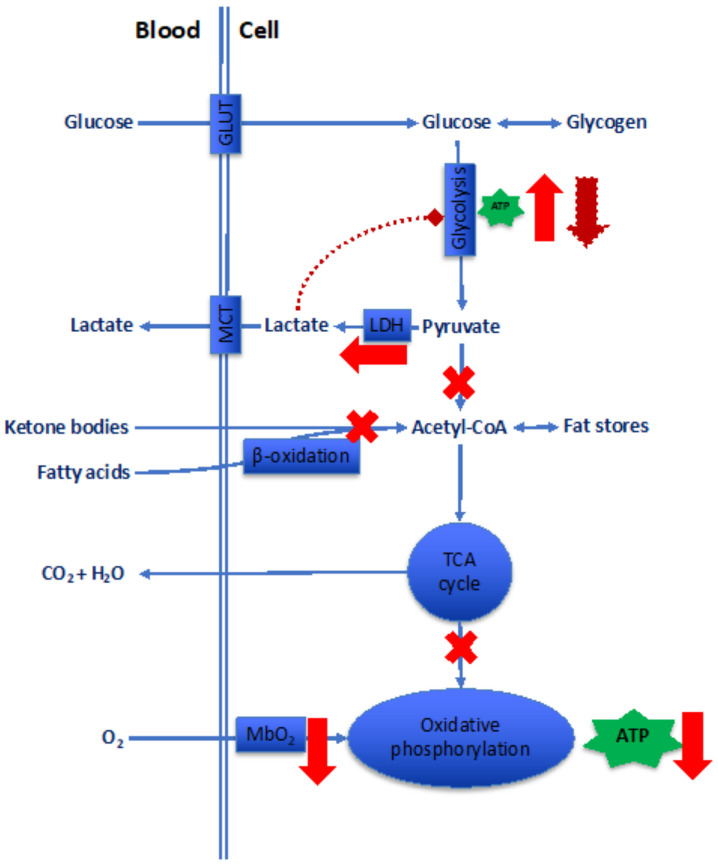
图5.ATP生成途径示意图
在常氧状态下,大部分ATP通过有氧方式生成,而氧化磷酸化过程(三羧酸循环[TCA循环]的后续步骤)对氧气(O₂)有严格需求。收缩组织中肌红蛋白(Mb)的存在有助于氧气的利用。对有氧ATP生成贡献最大的可氧化底物包括葡萄糖、脂肪酸,以及(在心脏和大脑中)酮体。
在急性低氧状态下(红色箭头和叉号标注),氧化磷酸化过程的氧气供应减少,有氧ATP生成随之下降。低氧会下调氧化磷酸化相关功能,导致三羧酸循环对乙酰辅酶A(acetyl-CoA)的代谢能力减弱;而乙酰辅酶A代谢受阻又会进一步抑制脂肪酸(以及心脏和大脑中酮体)的利用。与此同时,糖酵解过程被激活,通过葡萄糖无氧代谢短暂增加ATP生成,但这一过程会伴随乳酸合成增多,仅能部分代偿有氧ATP生成的减少。
在长期低氧状态下(棕色虚线标注),至少在收缩组织中,乳酸的过度生成无法通过单羧酸转运体(MCT)的成比例上调得到代偿(详见3.1.4 乳酸穿梭)。这会导致细胞内乳酸蓄积,而乳酸会抑制糖酵解通量,进而损害无氧ATP的生成。
低氧对骨骼肌线粒体体积和密度的影响存在争议,这主要是因为两种相互叠加的效应共同作用:一方面,运动训练本应增加线粒体体积;另一方面,低氧却会拮抗训练对线粒体产生的上述效应[74]。显然,低氧的持续时间、强度以及运动程度等变量均会对结果产生影响[75]。不过,动物研究表明,无论是急性还是慢性低氧,肌肉细胞色素氧化酶的活性都会显著下降,而这一酶活性降低会抑制线粒体的能量生成功能[76]。
在人类研究中,排除急性运动训练干扰的情况下,有报道显示夏尔巴人[77]和居住在低海拔地区的藏族人[78],其肌肉线粒体含量低于低海拔人群。此外,高海拔夏尔巴人肌肉中编码过氧化物酶体增殖物激活受体α(PPARα,一种脂肪酸代谢的转录调控因子)的基因表达下调,导致其脂肪酸氧化能力降低[79]。事实上,在针对低海拔人群进入高海拔环境的生理性低氧研究中,线粒体体积减少且氧化能力下降是一致观察到的结果[80]。
随着线粒体功能受损,糖酵解应成为为细胞提供无氧ATP的关键途径,这一过程即所谓的“糖酵解转换”或“瓦伯格效应”(Warburg effect)。瓦伯格效应最初在低氧癌细胞中被发现,指细胞代谢对糖酵解的依赖程度增加,以避开受损的线粒体功能[81]。研究发现,缺氧诱导因子-1α(HIF-1α)可激活瓦伯格效应[82],这为将糖酵解转换视为低氧应答提供了结构基础。尽管糖酵解产生生物能量的效率较低,但它确实能生成ATP——例如,人类红细胞(RBC)的能量供应完全依赖糖酵解。此外,高强度运动时的肌肉也会偶尔依赖无氧代谢供能,尽管其能量转换效率较低。
糖酵解过程会释放乳酸和氢离子(H⁺)。虽然高浓度氢离子在离体灌流心脏中仅会引发可逆性效应,但高浓度乳酸却会通过至少两种机制对氧气利用途径造成不可逆损伤:(a)乳酸-氢离子共转运体上调,导致线粒体内膜氢离子梯度耗散,进而损害磷酸化偶联效率;(b)乳酸诱导钙离子(Ca²⁺)瞬变幅度增大,使细胞内钙离子负荷增加,而将钙离子泵出细胞外所需的能量消耗也随之升高[83]。
细胞外排乳酸的限制因素将在下文讨论。被排出细胞的乳酸会通过肝脏的糖异生作用重新转化为葡萄糖,这一过程每生成1摩尔葡萄糖需消耗6摩尔ATP;而在糖酵解过程中,1摩尔葡萄糖转化为乳酸仅能产生2摩尔ATP。不过,肝脏进行糖异生(即科里循环,Cori cycle)所需的ATP实际上是通过有氧代谢生成的——科里循环的作用是代偿外周低氧细胞无氧代谢产生的副产物,而这一过程再次表明氧气是维持生命不可或缺的底物。
瓦伯格效应导致的细胞内乳酸蓄积,对细胞微环境的酸化作用远大于氧化磷酸化产物二氧化碳(CO₂)。细胞酸度升高会引发多种副作用,这些副作用通常被认为具有危害性。瓦伯格效应常见于癌症发生过程中,也存在于低氧肺部的所有组成细胞中,包括平滑肌细胞、内皮细胞和成纤维细胞[84]。针对人类肺动脉高压(PH)的研究[85]以及对肺动脉高压实验模型的荟萃分析[86]均证实:右心室底物利用模式的变化与疾病严重程度相关,且这种变化与瓦伯格效应的发生相符。
近年来,研究人员在帕金森病的体内和体外模型中,探究了促红细胞生成素(EPO)对线粒体功能的积极作用——在帕金森病模型中,氧化应激增强会导致线粒体功能障碍、神经元凋亡和细胞毒性[87,88]。结果显示,施用EPO虽无法恢复线粒体功能,但能加速糖酵解速率,从而部分恢复ATP水平,其作用方式与低氧适应过程类似[89]。然而,实际情况是,低氧开始后几天内EPO水平通常达到峰值,随后几周逐渐恢复至基线,这使得很难确定EPO在保护线粒体免受长期低氧影响及低氧适应过程中所发挥的作用。
综上,低氧相关的生物能量变化是有氧代谢向无氧代谢转换的必然结果,这些变化可能对低氧适应有益。但目前尚无方法能在不借助间接数学计算的情况下,直接在体内线粒体水平测量氧气输送情况,这使得我们无法准确评估生物能量变化对低氧适应的具体影响。此外,尽管已有假说提出[90],促红细胞生成素等多效性因子可能调控有氧和无氧能量生成,但这些因子的具体作用仍有待进一步研究。
3.1.4 乳酸穿梭
作为瓦伯格效应的产物,有氧代谢向无氧代谢的转换不仅会因能量生成受损对细胞造成负担,还会因乳酸过度生成产生不利影响。不过,通过促进乳酸向细胞外排出,至少可在一定程度上对抗乳酸的毒性作用。“乳酸穿梭假说”描述了糖酵解产生的乳酸在细胞内及细胞间的转运过程[91]。尽管乳酸常被视为无氧细胞代谢的毒性产物,也是导致肌肉疲劳的因素之一,但它实际上介导了多种生理过程,包括伤口修复与再生、星形胶质细胞-神经元协同作用、肝脏通过科里循环将乳酸-丙氨酸转化为葡萄糖,以及过氧化物酶体功能和精子发生[92]。
乳酸穿梭的关键因素是单羧酸转运体(MCT)——这是一个广泛表达的膜转运体家族,至少包含14种亚型,可介导乳酸、丙酮酸和酮体跨细胞膜转运[93]。目前研究最多的几种MCT亚型对乳酸的米氏常数(KM)存在差异[94]:其中,MCT2对乳酸的亲和力较高(KM=0.5-0.75 mM),而MCT1的亲和力较低(KM=3.5-10 mM)[95]。MCT4最初被认为是低亲和力转运体,但后续研究发现,它实际上是一种高亲和力的乳酸-氢离子同向转运体,能够逆显著浓度梯度将乳酸排出细胞[96]。
MCT在低氧环境中的作用,主要源于以下观察结果:MCT的高表达与肿瘤的侵袭性和恶性程度相关[97]。在三阴性乳腺癌中,MCT4表达增加与临床预后相关[98];而在胶质母细胞瘤中,MCT1上调会维持其糖酵解表型[99]。MCT1和MCT4对糖酵解型肿瘤的生长均至关重要[100],甚至有研究发现,MCT2能响应微环境的低氧和酸中毒,维持胶质母细胞瘤的高糖酵解通量[101]。这些发现为以MCT介导的乳酸转运为靶点治疗某些类型癌症提供了理论依据[102]。事实上,通过抑制MCT4减少乳酸排出,会导致细胞内酸中毒加剧,进而降低癌细胞存活率[103]。同样,靶向内皮细胞中的MCT1,可通过抑制乳酸诱导的HIF-1激活,从而抑制肿瘤血管生成[104]。此外,乳酸还可通过结合并稳定一种能触发细胞生长和血管生成信号的蛋白质,独立于HIF介导低氧应答[105]。
除肿瘤外,MCT在涉及免疫抑制、炎症、胰岛素抵抗和脑功能的疾病中也具有临床意义。在腓肠肌(红色肌纤维为主)中,几乎所有MCT亚型均在mRNA水平表达,且在低氧预处理和运动条件下,其表达调控模式存在差异[106]。长期低氧会诱导间充质干细胞中MCT4的表达,其分泌组会对心血管修复产生有害影响[107]。
从直觉上看,在生理性低氧状态下,MCT和乳酸脱氢酶的表达应会增加,以促进糖酵解产生的乳酸排出,从而防止细胞微环境酸化。然而,对暴露于海拔4500米/氧气浓度12.1%环境8周的大鼠研究发现,MCT的应答具有极强的组织特异性:心脏中MCT4表达增加34%,而跖肌中MCT4表达减少47%;同时,跖肌中MCT1表达也降低。这表明在低氧收缩组织中,MCT的作用相对有限[108]。
与之相反,在体外培养的人类脂肪细胞中,48小时低氧处理会使MCT1的mRNA和蛋白表达分别增加8.5倍和2.7倍,MCT4的mRNA表达增加14.3倍,但MCT4蛋白表达无变化[109]。在轻度低氧(海拔1390米/氧气浓度18.0%)环境下训练2周的纯血马,其MCT4蛋白水平和磷酸果糖激酶活性升高,而MCT1蛋白水平保持稳定[110]。暴露于更严重低氧环境(海拔3000米/氧气浓度14.7%)的马匹,MCT1蛋白无变化,但MCT4蛋白增加13%——这表明低氧至少可改善马匹的运动表现和骨骼肌糖酵解能力[111]。最后,在大鼠比目鱼肌和伸趾长肌中,高强度间歇训练(一种模拟低氧的模型)会上调MCT4、过氧化物酶体增殖物激活受体γ辅激活因子1α(PGC-1α)和HIF-1α的mRNA水平,但目前尚无相关蛋白水平的数据[112]。
上述数据似乎表明,MCT1对氧气变化不敏感,而MCT4的表达则依赖于氧气水平。
综上,目前针对“MCT通过促进体细胞乳酸排出来参与低氧适应”的靶向研究较为缺乏,因此尚无充分证据支持MCT在低氧适应中发挥重要作用。
3.2 毛细血管-细胞界面
3.2.1 一氧化氮
尽管一氧化氮(NO)在循环系统中的半衰期仅为数毫秒[113,114],但它是低氧状态下循环应答的关键介质——通过血管舒张作用恢复被低氧破坏的氧供需平衡,尤其在急性低氧场景中效果显著[115]。一氧化氮是血管张力和血压的主要调控因子[116]。此外,一氧化氮的其他重要功能还通过蛋白质亚硝基化实现,例如对缺氧诱导因子-1α(HIF-1α)的亚硝基化修饰:HIF-1α不仅会被低氧稳定,还会通过一氧化氮依赖性的S-亚硝基化作用维持稳定[117]。
一氧化氮是一种小分子自由基,由一氧化氮合酶(NOS)催化生成,反应中氧气是底物之一,具体反应式如下:
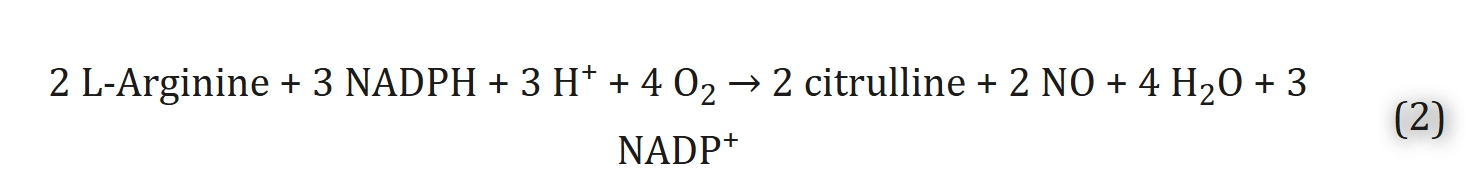
至少有三种不同的NOS亚型可催化上述反应,每种亚型由不同染色体编码:
1. 钙敏感性组成型神经型一氧化氮合酶(nNOS):主要分布于神经组织、平滑肌和骨骼肌中,其产生的一氧化氮大部分作为神经递质发挥作用;
2. 钙不敏感性诱导型一氧化氮合酶(iNOS):主要存在于免疫系统、心血管系统、血管壁和巨噬细胞中,可被炎症刺激和低氧诱导激活;
3.钙敏感性组成型内皮型一氧化氮合酶(eNOS):几乎在所有器官的内皮细胞中均有表达,存在基因多态性[118,119],其主要功能是介导平滑肌舒张。
低氧对NOS活性的调控,能够精细调节向细胞的氧气输送[120]。NOS对氧气的米氏常数(KM)是理解一氧化氮生成机制对氧气敏感性的关键。简而言之,大脑和内皮细胞中一氧化氮的合成速率与低氧的发生密切相关——这是因为NOS总活性对氧气的KM值(3.6-14.3 mmHg/0.5-1.9% O₂[121])接近组织预期的氧分压(PO₂),使得氧分压变化与一氧化氮生成动态关联。nNOS亚型对氧气的KM值为158 mmHg/20.8% O₂[122],理论上可响应氧分压变化,但由于微环境氧分压远低于其KM值,因此其应答能力有限。与之相反,eNOS对氧气具有高亲和力——在大脑和内皮细胞中,其对氧气的KM值分别为16.6 mmHg/2.2% O₂和5.5 mmHg/0.7% O₂[121]。这表明,除非在极端低氧条件下,否则eNOS所处环境的氧气浓度已能使酶达到饱和状态;因此,在非极端情况下,eNOS感知的氧分压变化预计与一氧化氮生成变化无紧密关联。
一氧化氮释放到血液后,会与至少三种成分建立平衡(图6):(1)以血浆中亚硝酸盐(NO₂⁻)和硝酸盐(NO₃⁻)形式存在的无机储存库;(2)与红细胞血红蛋白(Hb)结合形成亚硝基化血红蛋白或亚硝基硫醇化血红蛋白[123];(3)与平滑肌细胞中的可溶性鸟苷酸环化酶结合,生成环磷酸鸟苷(cGMP)——cGMP通过激活蛋白激酶G(PKG)并降低细胞内钙离子浓度,最终诱导平滑肌舒张[124]。

图6.平滑肌细胞中调控NO-cGMP通路的机制简化示意图
图中展示了四个关键区域:产生一氧化氮(NO)的内皮细胞(绿色)、以亚硝基化血红蛋白(HbNO)或亚硝基硫醇化血红蛋白(HbSNO)形式储存NO的红细胞(红色)、以无机亚硝酸盐和硝酸盐(NO₂⁻+NO₃⁻)形式储存NO的血浆,以及平滑肌细胞(浅蓝色)。在平滑肌细胞中,NO可促进肌浆网对钙离子([Ca²⁺])的摄取,使[Ca²⁺]水平降低,进而抑制肌球蛋白激酶活性,最终促进肌肉舒张。
循环系统中NO水平过高具有毒性——NO本身属于自由基,其与氧气(O₂)反应会生成过氧亚硝酸盐(一种极具危害性的活性氮物种)。这一过程会引发亚硝化应激,而亚硝化应激在心力衰竭[125]等多种疾病的病理机制中发挥重要作用。相反,当NO水平处于生理范围或略高于生理水平时,可通过多种途径对低氧状态下的机体起到保护作用。
除作为血管舒张剂以增强组织灌注和氧合这一主要功能外,NO还能恢复外周血单核细胞的黏附能力(低氧会显著降低该能力)。这一功能通过NO与影响组织重构及细胞外基质的机制相互作用实现,具体表现为:
1. 低氧诱导的内皮型一氧化氮合酶(eNOS)过表达,有助于恢复基础NO水平,进而通过环磷酸鸟苷(cGMP)依赖性蛋白激酶恢复细胞与基质的黏附[126]。这种黏附能力对维持正常稳态至关重要,而低氧会降低NO的生物利用度,从而破坏该稳态(下文将进一步阐述);
2. 低氧诱导的肺血流灌注过度会导致F-肌动蛋白应力纤维解聚,增加内皮细胞通透性(可能引发高海拔肺水肿),最终破坏内皮细胞稳定性;而通过NO供体或西地那非激活NO/cGMP通路,可逆转这一过程[127]。
NO与低氧的关联十分紧密:
- 在急性低氧情境下(数小时至数天),低氧会降低肺部NO水平并引发血管收缩,最终可能导致低氧性肺血管收缩[128,129,130]。对于高海拔肺水肿患者,吸入NO可改善动脉血氧饱和度,从而缓解症状[131];
- 与之相反,一项针对健康成年人的研究显示:在海拔4559米/氧气浓度12.0%的环境中居住1周,并通过饮食补充硝酸盐后,受试者唾液和呼出气体中的NO水平升高(表明肺部NO可利用度增加),但血流动力学指标、血氧饱和度及急性高原病(AMS)的发病情况均未得到改善[132]。
动物模型研究则得出了存在争议的结果:
- 一方面,生活在青藏高原海拔3000-5000米/氧气浓度14.7%-11.3%环境中的耐低氧哺乳动物——高原鼠兔(Ochotona curzoniae),其体内NOx(NO的代谢产物)水平低于低海拔对照组,表明高海拔环境会抑制NO生成[133];
- 另一方面,高海拔新生羔羊的NO功能强于低海拔对照组,且其肺组织中RhoA蛋白表达增加,这提示NO介导的血管舒张对维持肺血管阻力具有重要意义[134]。
不过,目前学界的普遍共识是:低氧开始后2小时内,肺和血浆中的NO水平会下降;到48小时时,NO水平倾向于恢复至基线;5天后则会进一步升高至基线以上[135]。有趣的是,褪黑素(一种由松果体分泌的多效性激素,具有抗氧化能力,且可促进缺氧诱导因子-1α(HIF-1α)表达[136])能抑制低氧大鼠体内神经型一氧化氮合酶(nNOS)、eNOS、诱导型一氧化氮合酶(iNOS)及硝基酪氨酸的低氧诱导性升高[137]。此外,在海拔3500米/氧气浓度13.8%的环境中急性适应7天的过程中,雌激素受体1和2似乎与NO-cGMP通路的激活相关——其表达水平与eNOS蛋白水平升高、血浆NO₂⁻+NO₃⁻水平升高及内源性eNOS抑制剂(不对称二甲基精氨酸)水平降低呈正相关[138]。
在长期低氧适应方面,一项对2012年前发表的32篇文献的综述显示:在所有高海拔人群中,藏族人肺、血浆和红细胞中的NO水平最高[135]。由此可见,NO生物利用度的提高似乎有助于人类适应高海拔环境,也是藏族人应对高海拔低氧的核心机制之一[139]。这一结论与以下研究结果相符:
- 在海拔3800米/氧气浓度13.3%环境中居住10个月的人群,其血浆中L-精氨酸水平升高[140];
- 在高海拔环境中补充L-精氨酸[141]或西地那非[142]具有益处。
值得注意的是,埃塞俄比亚阿姆哈拉高海拔原住民无红细胞增多症,且血红蛋白氧饱和度正常,但其体内NO和cGMP水平升高,这提示他们可能通过血管舒张实现适应——其脑循环对NO敏感,但对低氧不敏感[143]。此外,居住在海拔3500米的拉达克高海拔人群,其循环系统中雌激素受体2水平高于低海拔对照组,同时睾酮水平较低、eNOS蛋白水平及血浆NO₂⁻+NO₃⁻水平较高[144]。
综上,NO的代谢调控在低氧适应中发挥重要作用,尤其在长期(跨世代)低氧暴露场景下,这一作用更为关键。
3.2.2 毛细血管密度
克罗格毛细血管模型(Krogh capillary model)被广泛用于模拟圆柱形毛细血管内血氧分压(PO₂)的变化,以及描述向周围组织的氧气供应过程[145]。该模型有助于理解运动及脑血管功能障碍相关情境下的微循环机制[146]。尽管存在局限性,但该模型明确:组织氧合主要通过毛细血管募集调控,且氧气梯度变化(即低氧)、红细胞压积及红细胞转运时间的改变均会对组织氧合产生重要影响[147]。
由于低氧诱导的内皮细胞氧气梯度降低,直观上会显著影响氧气在循环-细胞界面的扩散;且对于肌肉、大脑等高耗氧组织而言,氧气从循环系统向细胞的高效转运可能是关键瓶颈。因此,下文将针对这些组织的低氧适应机制单独展开讨论。
肌肉组织中的毛细血管密度与低氧适应
近红外光谱技术的无创检测结果显示:低氧导致的氧气梯度下降会降低肌肉组织中的氧气扩散效率,但毛细血管密度的增加可对此起到代偿作用[148]。因此,提高毛细血管密度(或肌肉组织中的毛细血管/肌纤维比值,即血管适应),可通过匹配氧气输送与肌细胞变化的代谢需求,助力低氧适应[149]。
一项针对安第斯山脉原生豚鼠的开创性研究发现:其骨骼肌的毛细血管供应比低海拔饲养的豚鼠高30%。这表明,更高的氧化能力需要更丰富的毛细血管网络、更短的氧气扩散距离及更高的肌肉肌红蛋白浓度[150]。另一项针对海拔4200米/氧气浓度12.26%环境中原生安第斯骨顶鸡(Fulica americana peruviana)的研究也证实:与低海拔出生的个体相比,尽管安第斯骨顶鸡的肌纤维直径更小,但其所有肌肉组织的单位面积毛细血管数量及毛细血管/肌纤维比值均更高[151]。
然而,这些来自高海拔适应动物的研究结果,在人类中并未得到证实。对暴露于海拔5000米/氧气浓度11.3%环境8周的人类骨骼肌进行形态学研究发现:毛细血管/肌纤维比值的升高并非由血管生成增加(即毛细血管网络扩张)引起,而是由肌肉横截面积和肌纤维尺寸减小导致——这两者均与肌原纤维蛋白丢失相关[152]。此外,生化研究也发现:在海拔4500米/氧气浓度12.0%环境中暴露8周后,人类骨骼肌的血管生成不足,表现为血管内皮生长因子(VEGF)、flt-1及flk-1 mRNA等血管生成标志物水平降低[153]。类似趋势也出现在慢性心力衰竭患者中——其肌肉毛细血管密度和肌纤维面积均无显著变化[154]。
通过理论分析评估“毛细血管网络生长是否为肌肉组织低氧适应应答”时发现:当毛细血管/肌纤维比值超过2后,进一步增加该比值可能无法改善组织氧合[155]。因此,毛细血管密度的变化或许并非低氧适应的应答机制。这一看似违背直觉的结论,可通过克罗格模型的局限性得到解释:
- 核心问题在于,基于圆柱形结构的克罗格模型可能无法充分描述氧气向组织的输送过程,因为毛细血管并非组织氧气的唯一来源。实际上,小动脉也可能是重要的氧气供应源——磷光衰减技术对局部血管内和血管外PO₂的检测数据已证实这一点[156];
- 关于血管纵向梯度的研究一致表明:小动脉可通过氧气驱动的自分泌效应调控组织血流[157];且血液黏度、血液携氧特性及血红蛋白-氧气解离曲线斜率等变量,均需纳入考量才能完整描述氧气向组织的转运[158];
- 此外,毛细血管内红细胞转运时间和红细胞压积的局部异质性,也是匹配氧气供应与代谢需求变化的关键因素[159]。事实上,短期制动后毛细血管密度显著增加(扩散距离缩短),但并未显著改善运动中骨骼肌的氧气摄取效率[146]。
脑组织中的毛细血管密度与低氧适应
为应对氧气供应波动、维持正常功能,脑组织会启动特定机制:通过HIF-1α过表达驱动血管重构,激活下游基因(尤其是VEGF),进而增加毛细血管密度和脑血流量[160]。具体表现为:
- 在海拔4500米/氧气浓度12.0%环境中持续暴露28天,会导致毛细血管直径和长度增加,引发血管重构[161];
- 在海拔5900米/氧气浓度10.0%环境中暴露28天,会使CD34和PECAM-1等血管标志物水平升高[57]。
越来越多的证据表明:脑组织内的神经干细胞(位于脑内特定微环境中)可通过氧气依赖性调控机制调节脑毛细血管网络——轻度低氧可增强神经干细胞的增殖能力和多向分化潜能[162]。不过,HIF-1α在这一机制中的作用尚不完全明确:部分研究认为HIF-1α是必需的[163],而另一部分研究则发现其仅通过抑制凋亡、促进神经干细胞自我更新,来辅助信号传导通路[164]。其他可能参与该过程的机制还包括Wnt/β-连环蛋白信号通路[165]、钙调控的钙调神经磷酸酶-NFATc4通路[166]及活性氧(ROS)[167]。
此前研究发现:低氧可调控内皮细胞中VEGF基因的表达,通过自分泌方式影响内皮细胞的通透性和增殖[168]。此后,大量研究围绕“低氧驱动的血管生成调控”展开,发现多种细胞类型中存在数量不断增加的调控通路[169]。如今,低氧诱导的VEGF过表达已被公认为恶性肿瘤扩张和血管功能调控中的关键因素[170],而HIF作为“血管生成总开关”,可对VEGF表达进行调控[171,172]。事实上,在HIF的多个靶标[49]中,VEGF及多种血管生成标志物与低氧适应的关联最为密切[173,174]。
综上,尽管在肌肉、大脑等高耗氧组织中,毛细血管密度的变化似乎并非低氧适应的关键因素,但在肿瘤组织(尤其是实体瘤)中情况则有所不同:耗氧型恶性肿瘤细胞的生长速度超过毛细血管网络的生长速度,导致毛细血管/细胞比值降低,最终引发局部低氧。
3.3 动脉血-毛细血管界面
3.3.1 红细胞生成
促红细胞生成素(EPO)是一种由肾脏间质成纤维细胞合成的糖蛋白,长期以来被认为是低氧状态下刺激骨髓红细胞生成的关键因子[175,176]。这一机制的发现与HIF的发现密切相关——氧气缺乏对HIF稳定性的调控,以及HIF对EPO生成的调控,是理解氧气传感器多效性机制的核心[177]。
EPO生成后,会与多种组织(包括骨髓和神经元)细胞表面表达的EPO受体结合,激活Janus激酶2(JAK-2),进而启动信号转导与转录激活因子(STAT)、磷脂酰肌醇3-激酶/蛋白激酶B(PI3K/Akt)及细胞外信号调节激酶(Erk)通路,最终产生包括红细胞生成在内的多种生物学效应[178]。
大量研究表明:EPO对低氧的应答具有时效性——低氧开始后数小时内EPO水平即升高,1-2天达到峰值,随后在1-2周内逐渐恢复至基线;而红细胞总量则在EPO峰值后缓慢升高,且这一过程会持续1周左右[179]。值得注意的是,尽管低氧开始1周后EPO水平已恢复至基线,但在高海拔环境中,红细胞生成的刺激信号至少会持续10个月[180]。
这一机制中,可溶性EPO受体(EPO的内源性拮抗剂)发挥重要作用:其对低氧的应答模式与EPO一致——在海拔4340米/氧气浓度12.4%环境中,可溶性EPO受体水平降低19%,且在至少72小时内维持在基线以下,随后缓慢恢复[181]。这表明,EPO与EPO受体的比值是理解EPO与红细胞总量长期关系的关键。
通过对高海拔常住人群的比较分析,可进一步深入理解“低氧→HIF→EPO→红细胞生成”通路的机制:
- 藏族人无明显红细胞增多症[182],且高海拔与低海拔藏族人的EPO水平无差异[183];与藏族人存在种族关联的拉达克高海拔人群,其EPO水平也较低[184];
- 居住在相似海拔的埃塞俄比亚阿姆哈拉人,与低海拔人群相比同样无红细胞增多症[185];
- 与之相反,安第斯高海拔人群以红细胞增多症著称[186],且多数情况下红细胞增多症是慢性高原病(CMS)的先兆。尽管人们预期其EPO水平会更高,但实际情况并非如此:在海拔4340米/氧气浓度12.0%环境中居住的南美洲人群中,循环EPO水平并非CMS的鉴别因素,而EPO与EPO受体的比值可能是导致红细胞生成过度的关键[187];此外,安第斯高海拔人群中,血红蛋白水平高低不同者的EPO水平仅存在微小差异[188]。
增加红细胞生成或许是最早被确认的低氧适应生理应答之一[189]。这一应答随后被证实由EPO过度生成介导[190],且因能成比例提高组织氧气输送量,被认为是适应性应答[191]。然而,红细胞增多症对血液氧气转运的有害影响,主要源于血液黏度升高导致的剪切速率增加——而剪切速率是决定局部血流的关键因素[192]。
红细胞压积是影响血液黏度的最重要单一因素,此外还包括纤维蛋白原、红细胞变形能力及聚集性等[193]。在健康人群中,红细胞压积在正常范围(33%-45%)内波动时,脑血流量仅发生轻微变化;但超出该范围后,红细胞压积升高会导致血流量降低[194]。
综上,适应高海拔的人群表现出红细胞生成应答减弱,而未适应人群则常因红细胞生成过度出现不良临床特征——这一现象提示,红细胞生成或许并非低氧的适应性应答。
3.3.2 铁代谢
红细胞生成增加必然导致铁负荷加重。如图7所示,血红蛋白(Hb)合成至少需要两个关键因素:一是前文讨论的低氧和EPO驱动的刺激信号,二是铁供应——因此,铁供应与低氧的强度和持续时间密切相关[195]。
在生理性低氧状态下,合成血红蛋白对铁的需求增加,这一需求通过两种途径满足:一是增加肠道铁吸收,二是调动肝脏(铁储存器官)中的铁储备。
铁转运体 ferroportin( ferroportin)是肠上皮细胞和肝细胞基底外侧膜上的主要铁转运蛋白,其活性受铁调素(hepcidin)的严格调控:铁调素可促进ferroportin内化,从而降低铁向循环系统的输出能力[196,197]。在急性低氧状态下(数天至数周),低氧会下调铁调素水平,从而促进铁进入血液,为红细胞生成提供原料[198]。Figure 7.
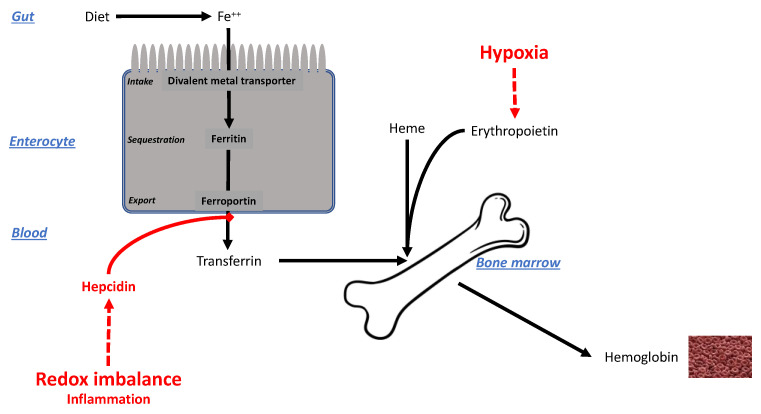
图7.铁代谢机制及低氧与炎症的影响示意图
然而,在病理性低氧状态下,情况则有所不同。慢性阻塞性肺疾病(COPD)是一种气流受限疾病,随着病程进展,患者的内皮功能会显著下降,并伴随低氧症状[199]。在COPD患者中,40%-50%会通过生理性红细胞生成适应低氧环境,但仍有5%-30%会出现缺铁性贫血[200,201]。值得注意的是,贫血已被证实是预测患者预后不良的风险因素[200,202,203]。急性呼吸窘迫综合征(ARDS)常以严重低氧为特征[204],而贫血是该疾病公认的合并症。
生理性与病理性低氧引发的应答差异,被认为与铁调素(hepcidin)的调控机制不同有关,而这种调控差异由氧化还原失衡驱动。在ARDS和COPD患者中,氧化还原失衡程度更高,且在强烈炎症刺激(如C反应蛋白水平升高)的情况下,氧化还原失衡会成为调控红细胞生成的额外因素[205]。此外,新冠肺炎(COVID-19)患者(尤其是疫情早期患者)常表现出与ARDS患者类似的严重低氧症状。尽管目前关于COVID-19在呼吸衰竭发展、肺顺应性及内皮炎症方面是否与ARDS相似仍存在争议[206],但有研究表明,COVID-19患者的氧化还原失衡程度和细胞因子风暴强度均高于ARDS患者[205]。铁驱动的炎症会选择性刺激鞘脂合成[207],其中1-磷酸鞘氨醇(一种COVID-19综合征中的关键介质)的合成会显著增加[208]。
综上,至少在无炎症因素的情况下,机体对生理性低氧的铁代谢应答会促进红细胞生成,但这一过程未必是有利因素。因此,在生理性低氧状态下,铁调素的下调可被视为一种适应不良表现。
3.3.3 血红蛋白氧亲和力
血液的携氧能力主要取决于血红蛋白(Hb)浓度(详见其他章节)和血红蛋白-氧气(Hb-O₂)亲和力。在特定血氧分压(PO₂)下,Hb-O₂饱和度由以下因素决定:pH值、二氧化碳分压(PCO₂)、红细胞内2,3-二磷酸甘油酸(DPG,一种糖酵解中间产物,可与Hb的T态结合并调控Hb功能)水平、温度,以及碳氧血红蛋白(与一氧化碳结合的Hb)水平[209]。P50(即Hb-O₂饱和度达到50%时的PO₂)是评估Hb-O₂亲和力的重要指标。
在低海拔至中海拔范围内,Hb-O₂亲和力降低(即P50升高)通常被认为是有利的——因为它能通过降低静脉Hb-O₂饱和度来增大动静脉氧分压差[210]。根据菲克方程(该方程将氧耗量与心输出量、动静脉氧含量差相关联),Hb-O₂亲和力降低(或P50升高)可能带来两种益处:一是改善组织氧合(P50每变化1 mmHg,静脉PO₂相应变化+3.2 mmHg);二是减轻循环负荷(P50每变化1 mmHg,心输出量相应降低5.8%)。
与之相反,在极高海拔环境中,高Hb-O₂亲和力(即P50降低)更有利于组织供氧,因为它能补偿动脉Hb饱和度的下降[211]。研究表明,在海拔6450米处,动脉pH值每变化0.1个单位,Hb-O₂饱和度最多可变化5%[212]。因此,在中海拔环境中,碱中毒(其成因将在下文探讨)会升高Hb-O₂亲和力、降低P50,从而产生有害影响;而DPG水平升高则会通过相反机制发挥有利作用。如表2所示,在海拔6450米处,上述两种因素的变化几乎完全相互抵消,使得P50值基本保持不变,这与早期假说一致[213]。
表2. 不同海拔下白种人与夏尔巴人的氧气运输特征
除P50外,所有数据均来自[212]。P50的计算基于[209]中报告的公式,假设碳氧血红蛋白含量为0且温度为37°C。
人群 | 海拔 / 氧气浓度 | 动脉 pH 值 | 动脉 PCO₂(mmHg) | 动脉 PO₂(mmHg) | 血红蛋白浓度(g/L) | DPG/Hb 比值 | P50(mmHg) |
白种人 | 0 米 / 21.0% | 7.38 | 40 | 95 | 160 | 0.80 | 27.9 |
3400 米 / 13.9% | 7.46 | 22 | 51 | 170 | 1.03 | 27.4 | |
5050 米 / 11.3% | 7.48 | 21 | 44 | 191 | 1.04 | 26.9 | |
6450 米 / 9.3% | 7.50 | 19 | 34 | 201 | 1.36 | 28.8 | |
夏尔巴人 | 3400 米 / 13.9% | 7.46 | 29 | 53 | 169 | 1.01 | 27.5 |
5050 米 / 11.3% | 7.45 | 27 | 42 | 186 | 1.04 | 27.9 | |
6450 米 / 9.3% | 7.45 | 22 | 33 | 未检测(NA) | 1.20 | 29.0 |
然而,需要强调的是,要全面理解高海拔环境下血液氧运输的意义,还需考虑两个较少被分析的因素:
1. 温度波动:在极端环境中,温度波动的影响不容忽视——温度每变化1°C,P50约变化1.5 mmHg[214],这一数值与表2中所示的P50变化幅度相当;
2. 高海拔诱导的红细胞生成刺激:该刺激会使循环中“年轻”红细胞(新合成的红细胞)的比例增加[215]。骨髓生成的年轻红细胞其DPG/Hb比值高于“衰老”红细胞(0.96±0.13 vs. 0.57±0.13 摩尔/摩尔)[216],对应的P50差异为4.3 mmHg(从29.1 mmHg降至24.8 mmHg),远大于表2中报告的P50变化幅度。
因此,即使血液中DPG/Hb平均值正常,不同红细胞的氧亲和力分布范围也可能很广。遗憾的是,目前无法确定哪种类型的红细胞会主动参与毛细血管中的氧交换,或更倾向于进入动静脉分流。但有假说认为:若柔韧性较好的红细胞能通过毛细血管的弯曲路径,则年轻红细胞(平均 corpuscular 血红蛋白浓度较低、DPG/Hb比值较高,故氧亲和力较低[215])可能会主动释放氧气;而衰老红细胞(平均 corpuscular 血红蛋白浓度较高,故黏性更大、DPG/Hb比值较低、氧亲和力较高)则可能相反。需注意的是,由于缺乏相关数据支持,这一讨论仍具有较强的推测性。
综上,尽管Hb-O₂亲和力的变化理论上可能参与低氧适应,但实际观察到的变化幅度极小(若存在变化),因此不能认为其在低氧适应中发挥关键作用。
3.4 肺泡-动脉血界面与循环应答
3.4.1 心输出量
急性低氧会导致心输出量增加,其主要机制是通过代偿性心率加快,以向组织输送更多氧气[217]。这一应答的核心是肾上腺素能G蛋白系统——儿茶酚胺水平升高和肾上腺素能纤维活性增强均能证明这一点[218]。
关于每搏输出量变化对高海拔心输出量的影响,目前仍存在争议:多数研究发现,急性低氧暴露后每搏输出量未发生显著变化[219]。近期在模拟高海拔的低氧舱中对健康人群的研究发现,心输出量增加的另一机制是全身血管阻力降低[220];此外,外周小动脉扩张[221]也有助于维持组织氧供需平衡[222]。尽管存在上述代偿机制,多项研究仍观察到高海拔环境下每搏输出量下降,其潜在原因可能是冠状动脉PO₂降低导致的收缩功能受损[223]。同时,由于基础心输出量已升高,机体在运动时通过增加心输出量来满足氧需求的能力会下降[224]。
相比之下,持续性低氧对心输出量的影响较小。长期低氧会导致:β-肾上腺素能受体下调[225],毒蕈碱受体上调,抑制性和刺激性G蛋白的表达与活性改变,以及腺苷酸环化酶系统应答减弱[218]。此外,心肌PO₂相关的局部机制可能调控心脏变时功能[226],通过减少代偿性心动过速进一步降低氧耗量。
综上,急性低氧状态下心输出量的变化(由心率和每搏输出量共同决定)无法维持长期低氧适应,也并非低氧适应的关键因素。
3.4.2 红细胞肺毛细血管转运时间
红细胞在毛细血管中可能没有足够时间完全结合氧气。尽管血红蛋白与氧气的结合反应速率极快(<10毫秒即可完成[227]),远快于氧气扩散速率,但在肺内氧气扩散过程中,若忽略氧气扩散系数、温度、流体黏度、膜化学性质及肺气体交换表面积的影响,仍存在以下三个限制因素:(a)肺泡-动脉PO₂差;(b)血气屏障厚度;(c)红细胞毛细血管转运时间。急性和慢性低氧均可能对这些因素产生显著影响。
1. 肺泡-动脉PO₂差
肺泡-动脉PO₂差通过通气/血流(V/Q)比值评估。低氧会破坏V/Q平衡(尤其是急性低氧状态下):相对通气不足会导致V/Q失衡,但肺血管收缩可通过将血流从通气不良区域导向通气良好区域来代偿,从而改善氧合。目前认为,肺血管收缩是调节V/Q平衡的主要主动机制。近期开发的计算模型总结了该领域的研究进展——该模型预测,低氧性肺血管收缩可通过改善V/Q平衡来匹配血流与通气、使区域肺泡-毛细血管氧通量均匀化,并增加循环中的氧气负荷[228]。
2. 血气屏障厚度
基底层(由上皮细胞形成的细胞外基质层,可作为细胞附着位点)可能是肺血气屏障中的主要阻力成分,其结构需在两个需求间平衡:一是提供机械阻力以抵抗过高压力,二是促进氧气穿过肺泡屏障扩散[229]。在与低氧相关的肺部疾病(如肺动脉高压(PH)引发的肺毛细血管应力衰竭导致的低氧性肺水肿[230],以及COPD[231])中,这种平衡会被打破。
对暴露于海拔5900米/氧气浓度10.0%环境2周的大鼠研究发现,其肺毛细血管基底层厚度几乎增加一倍;而磷酸二酯酶5抑制剂可有效阻止这一增厚过程,同时还能改善与肺动脉高压和右心衰竭相关的其他指标[232]。此外,研究证实,肺损伤后肺泡II型细胞中的缺氧诱导因子-1α(HIF-1α)通路会被激活,在肺修复过程中促进细胞增殖和扩散[233]。
3. 红细胞转运时间与肺毛细血管重构
急性低氧状态下心输出量增加会导致红细胞转运时间成比例缩短。正常情况下,尽管肺内红细胞转运时间分布范围较广,但肺泡毛细血管中红细胞的平均转运时间约为0.75秒[234]。对兔离体肺的研究发现,即使血流量增加6倍,氧合也未受到限制[235]。
研究人员通过菲克扩散方程进行简单边界分析,探讨了红细胞转运时间缩短与红细胞与肺泡气体完全平衡之间的矛盾,并估算了红细胞氧气扩散的外部阻力[236,237]。在低海拔环境中,由于肺泡-动脉PO₂差较大,血红蛋白结合氧气的速率并非关键因素;但在高海拔环境中,肺泡氧气浓度低会限制氧气扩散,可能导致红细胞无法与肺泡氧气充分平衡。例如,在海拔3840米/氧气浓度13.2%环境下运动时,红细胞与肺泡氧气无法完全平衡,部分原因就是红细胞转运时间缩短[238]。
持续性低氧会引发肺重构,这是肺动脉高压发生的基础[239]。该过程涉及表达跨膜酪氨酸激酶受体c-kit的骨髓来源祖细胞的动员[240,241]。在“10%氧气、持续2周低氧”的大鼠模型中,肺内c-kit阳性细胞数量增加10倍,同时伴随显著的血管肌化、肺毛细血管大小重新分布(小毛细血管(<50微米)数量大幅增加,大毛细血管数量无变化[242])。即使低氧时间延长一倍,这种重构模式也基本保持不变,表明其可能是低氧适应的一部分[243]。然而,目前尚无法确定肺毛细血管大小变化对红细胞转运时间的影响,以及肺泡-动脉血界面在低氧适应中的具体作用。
此外,上述关于肺内红细胞氧结合的讨论,在反向应用于外周组织的氧释放过程时同样适用,但氧释放速率比氧结合速率慢两个数量级[237],这使得情况更为复杂。在以下情况下,红细胞氧释放可能会出现问题:转运时间缩短、内皮PO₂梯度降低、红细胞生成过多导致血液黏度改变、Hb-O₂亲和力变化、酸碱状态改变等。但目前关于这一主题的研究数据较少。不过,有分析表明,氧气扩散的阻力并非来自红细胞膜,而是来自红细胞表面的不流动层[237]。
综上,红细胞肺毛细血管转运时间(外周毛细血管转运时间的影响可能较小)可能是决定机体对急性和慢性低氧应答的重要因素。但要明确这一现象在低氧适应中的意义,还需对肺毛细血管网络的再生能力开展更详细的研究。
3.5 大气-肺泡界面与通气应答
3.5.1 颈动脉体
随着低氧程度逐渐加重,通气频率会随之加快,以促进肺泡内的血液氧合。颈动脉体(CB)是位于颈总动脉分叉处外膜的细胞群,作为主要的化学感受器,它能感知氧气不足并启动这一反射[244]。一系列机制共同保障颈动脉体I型球细胞实现氧气感知功能,其中核心机制之一是对氧敏感性钾离子(K⁺)通道的抑制[245]:钾离子通道受抑会导致细胞去极化,使钙离子(Ca²⁺)内流增加,进而促使含有多巴胺、乙酰胆碱、三磷酸腺苷(ATP)及儿茶酚胺等神经递质的细胞内囊泡释放[246],最终引起通气频率升高[247]。该机制的正常运作需要功能完整的电子传递链,且线粒体复合体I与复合体IV之间需保持完整的偶联状态[248]。线粒体复合体IV活性下调会导致还原型烟酰胺腺嘌呤二核苷酸(NADH)和活性氧(ROS)生成增加,而这两种物质会对膜离子通道活性产生负向调节作用[249,250]。
此外,还有其他机制可能影响颈动脉体球细胞的氧气感知功能。首先,一氧化氮(NO)、一氧化碳(CO)、硫化氢(H₂S)等非囊泡储存型气体信使的释放,会通过抑制颈动脉体兴奋、减弱通气应答来拮抗低氧反应[251]。与之相反,低浓度(<0.5 IU/mL)的促红细胞生成素(EPO)能增强颈动脉体对低氧的应答[252]。事实上,促红细胞生成素正逐渐成为一种有效的预防性治疗手段,不仅可用于治疗急性高原病(AMS),还能缓解新型冠状病毒(SARS-CoV-2)感染引发的多种急性呼吸及非呼吸症状[253]。
显然,目前对颈动脉体氧气感知功能的潜在机制仍需进一步深入研究,才能解释近年来的一些观察结果。例如,裸鼹鼠对低氧的颈动脉体敏感性显著降低,尽管其颈动脉体体积更大、球细胞数量多于小鼠,但低氧通气应答却明显减弱,这可能与血红素加氧酶-1阻断气体信使信号传导有关[254]。此外,高海拔鹿鼠的通气敏感性和颈动脉体生长均有所减弱,这可能与其缺氧诱导因子-2α(HIF-2α)的基因变异相关[255]。最后,瞬时受体电位锚蛋白1(TRPA1)是一种广泛表达于三叉神经和迷走神经的阳离子通道,它能感知吸入气体中的刺激性物质,且可被轻度低氧(海拔4000-2500米/氧气浓度13.0%-15.0%)激活,但对重度低氧(海拔5900米/氧气浓度10.0%)无应答[256]。有趣的是,SARS-CoV-2对颈动脉体的感染可能是新冠肺炎(COVID-19)患者出现“沉默性低氧血症”的原因,这提示或许可将颈动脉体激活剂用作COVID-19患者的呼吸兴奋剂[257]。原则上,颈动脉体的上述氧气感知机制也存在于主动脉体、星形胶质细胞等其他氧气传感器中[258]。
综上,颈动脉体对低氧的应答是一种已被明确且潜在影响显著的模式:适度增加通气频率可能具有保护作用,有助于抵消低氧的影响;但过度增加通气则可能表现为适应不良,例如可能引发过度通气导致的碱中毒,这一点将在下一小节中详细探讨。
3.5.2 过度通气与碱中毒
在低氧引发的过度通气作用下,肺部会过度排出二氧化碳(CO₂),导致动脉血二氧化碳分压(PCO₂)降低,进而引发碱血症。肾脏会通过代谢调节(使碱剩余值变为负值)对碱中毒进行部分代偿。研究发现,在海拔6450米/氧气浓度9.3%的环境中,未适应的白种人与适应程度更好的夏尔巴人表现出不同的生理模式[212]。尽管夏尔巴人长期(或许已达数千年[259])生活在海拔4000-5000米/氧气浓度12.9%-11.3%的环境中,但很难认为他们能完全适应6450米的海拔;不过,无论训练程度如何,他们对6450米海拔的适应程度几乎必然优于白种人。
尽管白种人与夏尔巴人的动脉血氧分压(PO₂)和血氧饱和度相同(图8),但白种人的动脉血二氧化碳分压更低,这可能是因为夏尔巴人对低氧的呼吸敏感性较弱。研究发现,高海拔原住民存在不可逆的呼吸驱动减弱现象[260],这一特征在高海拔早产儿[261]和肥胖低通气综合征患者[262]中也得到了证实,且与所谓的“快乐性低氧血症”(即患者虽有低氧但无明显不适)的发生相关[263]。对于暴露于极高海拔的夏尔巴人而言,呼吸驱动减弱具有保护作用:它可减少对过度通气的需求,从而减轻碱中毒程度。值得注意的是,白种人和夏尔巴人的碱剩余值均为-6 mEq/L,而完全代偿碱中毒所需的碱剩余值应为-10 mEq/L,这表明两者的代谢代偿程度相同。但与白种人相比,夏尔巴人的通气量更低,因此动脉血pH值也更低。
看似微小的pH值变化实则影响显著:正常pH值为7.4时,氢离子浓度[H⁺]为3.98×10⁻⁸;当pH值升至7.45时,[H⁺]降至3.54×10⁻⁸(较初始值降低10%);当pH值升至7.5时,[H⁺]进一步降至3.16×10⁻⁸(较初始值降低20%)。急性高原病患者的碱中毒程度更为严重,pH值可达7.57-7.63,此时[H⁺]为2.51×10⁻⁸,仅为正常水平的63%(降低37%)。
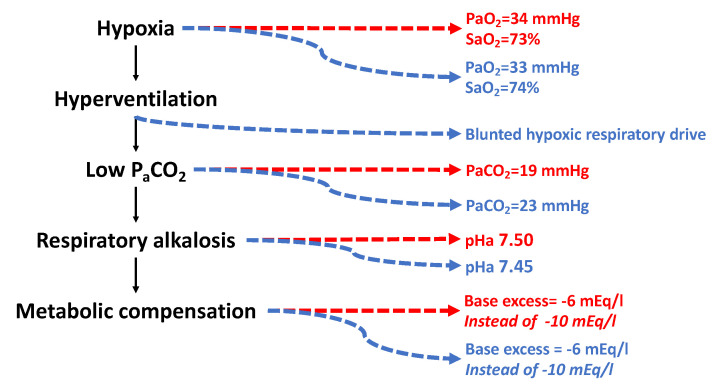
图8. 未适应的白种人(红色)与适应的夏尔巴人(蓝色)在海拔6450米/氧气浓度9.3%环境下的呼吸调节
数据来源:[212,264]
由此可见,通过过度通气纠正低氧血症可能并非理想选择,因为这会引发碱中毒。目前,持续碱中毒对脑功能的影响尚未完全阐明,但高血浆pH值可能至少会导致两种不良后果:(a)促进钙离子与白蛋白结合,在总钙水平不变的情况下降低血中离子钙浓度,使神经元和肌肉处于易激惹状态(手足抽搐);(b)导致脑血管收缩[265],进而损害脑功能。
关于高海拔环境下认知功能受损的直接研究数据较为有限,但两项荟萃分析均表明低氧会对认知产生负面影响[266,267],这一结论在“低海拔人群暴露于3269米海拔3天”的研究中得到了进一步证实[268]。目前,高海拔常住人群的认知功能受低氧影响已成为共识[269,270,271,272],因此有研究建议在高海拔地区的学校中补充氧气,以预防学习过程受到影响[24]。
综上,从二氧化碳流失引发碱中毒的角度来看,过度通气是对低氧的过度应答,可能对健康产生不利影响,符合“适应不良”的定义。
4 适应与适应不良
4.1 低海拔人群的高海拔适应能力
现有文献数据大多来自低氧暴露初期(数天至数周内),主要关注急性高原病的发生等适应不良表现;而关于长期(数月至数年)低氧暴露影响的数据相对较少,而这类数据恰恰能为低海拔人群的高海拔适应能力提供线索。
南极高原科考人员均为健康且体能良好的个体,他们经过专门训练,可在相当于海拔3800米/氧气浓度13.2%的环境中停留长达10个月。该环境排除了寒冷、海拔变化等干扰因素,唯一可能的例外是昼夜节律紊乱[180]。研究发现,这些科考人员的酸碱平衡状态在10个月内持续维持在低氧暴露后的改变状态,未出现明显恢复[180]。值得注意的是,除非极性代谢组在低氧暴露6个月后缓慢向基线水平恢复外[140],其他代谢组学指标在整个暴露期间均无显著变化。同样,活性氧、氧化应激生物标志物、一氧化氮及促炎细胞因子等与氧化还原失衡相关的指标,在低氧暴露第20天达到峰值后,始终未恢复至基线水平[67]。这些数据表明,至少在1年内,低海拔出生的白种人缺乏对高海拔低氧的适应能力。
低海拔人群长期居住在高海拔地区(如移居西藏的汉族人),慢性高原病(CMS)的发病率可高达18%[273]。因此,据目前所知,现有数据表明,即使暴露于低氧环境数年,低海拔人群仍无法适应这一挑战。
4.2 世代传承的低氧适应
在安第斯山脉、东南亚及埃塞俄比亚地区,约有8000万人长期居住在海拔>2500米的环境中[1]。这些人群对高海拔低氧形成了不同的适应模式:
- 安第斯人群:主要通过血液系统适应[274],其血红蛋白(Hb)水平升高,使血液能携带更多氧气,但慢性高原病发病率较高——成年男性发病率为16%,且随年龄增长而上升,50岁时可达30%[275],这表明其低氧适应能力较弱。
- 藏族人群:主要通过呼吸系统适应[274],表现为潮气量增大、呼吸频率加快,且血浆中亚硝酸盐(NO₂⁻)与硝酸盐(NO₃⁻)(一氧化氮储存能力的生物标志物)水平较高,因此藏族原住民中慢性高原病的发病率较低[273,276]。
- 埃塞俄比亚阿姆哈拉高海拔人群:慢性高原病发病率同样较低[277],同时表现为红细胞生成减少、血浆NO₂⁻+NO₃⁻及环磷酸鸟苷(cGMP)水平升高、舒张压降低。与移居高海拔的低海拔人群相比,该人群对低氧的血管舒张反应更为显著,而后者则表现出更强的红细胞生成应答[278]。
此外,吉尔吉斯通勤人群虽不属于典型高海拔人群,但具有研究价值:该人群中14%-20%存在高海拔肺动脉高压(PH)迹象,且对急性低氧的高应答者比例高于正常水平[279]。一项病例对照研究发现,健康的吉尔吉斯高海拔人群中存在特定遗传特征,可区分出会发展为肺动脉高压的低氧高应答者[280]。研究指出[280],与其他高海拔人群相比,吉尔吉斯通勤人群的适应模式具有独特性:除肺动脉高压外,他们无其他慢性低氧相关特征;而安第斯人群常伴有红细胞增多症(可能产生混杂效应),藏族人群则几乎无肺动脉高压表现。
因此,我们有理由认为,在喜马拉雅山脉出生并居住的高海拔人群(藏族、夏尔巴人,拉达克人程度稍弱)已“适应”高海拔环境——他们的高海拔相关疾病发病率更低,红细胞生成应答更弱。与之相反,安第斯人群(艾马拉人、克丘亚人)表现出过度的红细胞生成应答,红细胞压积超过50%,且因血液黏度升高而面临更高的器官功能障碍风险[259,281,282]。
有一种假说认为,藏族与安第斯人群的适应差异源于高海拔居住时间的不同:藏族在高海拔生活的历史约为3万年,有足够时间形成遗传适应;而安第斯人群的高海拔居住史仅约1万年[283]。不过,17世纪安第斯地区发生的大规模移民(打破了人群遗传稳定性),与喜马拉雅地区相对保守的人口分布形成对比,这使得上述时间差异的解释更为复杂。
4.3 高海拔人群的低海拔适应能力
尽管不常见,但在某些情况下(多因政治原因),高海拔适应人群被迫离开故土,前往海拔更低、环境更适宜的地区生活。数千名在高海拔出生但移居至低海拔地区的藏族难民,为研究高海拔适应过程的可逆性提供了独特契机——在低海拔环境中,低氧刺激消失,个体暴露于相对高氧状态。
图9(萨马贾等人,未发表数据)显示了移居至海拔<500米/氧气浓度>20.1%地区的藏族人的血液血红蛋白浓度,以及同期居住在该地区的印度血统人群(对照组)的血红蛋白水平。显然,低氧刺激的缺失会显著抑制红细胞生成,至少男性面临贫血风险。另一项大规模研究发现,长期居住在低海拔地区的藏族人,其红细胞计数、红细胞压积和血红蛋白水平均低于居住在高海拔的藏族人[183]。
还有一项研究进一步指出,与汉族人相比,居住在低海拔的藏族人血红蛋白浓度更低,同时表现为分钟通气量更高,且对急性(数分钟)和持续性(8小时)低氧的肺血管应答更弱[284]。该研究还发现,藏族人外周血淋巴细胞中缺氧诱导因子(HIF)调控基因的低氧诱导水平更低;与汉族人相比,藏族人的EPAS1和EGLN1基因型与促红细胞生成素(EPO)的低氧诱导水平存在显著相关性,这表明藏族人对低氧刺激的应答强度较弱[284]。
这些证据共同表明,高海拔适应人群在进入相对高氧的低海拔环境后,会产生相应的生理应答。但目前尚不清楚这种对相对高氧环境的血液系统应答是否具有危害性。
.
图9藏族与印度男性(分别为37人和56人)在海拔<500米/氧气浓度>20.1%环境下的血液血红蛋白浓度
藏族人群的血红蛋白(Hb)水平更低(p<0.0001,Student's t检验)。绿色线条代表95%置信区间。
5. 未来展望:组学研究
5.1 基因组学
不同人群对高海拔的应答存在显著差异,这为全基因组关联研究(GWAS)提供了独特契机,有助于识别决定全球不同人群对低氧应答差异的基因模式。在三个高海拔人群中,似乎有超过1000个基因可能参与低氧应答过程[285]。这些基因与神经、淋巴和心血管系统的发育相关,同时也涉及细胞生长、存活与死亡调控;不过,仅有4个基因(PIK3CB、HLA-DQB1、CNTNAP2和DLG2)在所有人群中均发生共同改变[285]。此类研究结果表明,低氧适应具有多基因特性,且会影响多个生化通路,而每个生化通路又会进一步影响多种生理应答。
尽管已有大量研究证实,部分生理机制(红细胞稳态、氧气运输、循环系统调节及氧气感知)是影响低氧适应的关键通路,但其他全基因组关联研究仍无法明确某些被认为与红细胞生成过多相关的基因所发挥的作用[286]。这一现象凸显了全基因组关联研究的一个主要局限性:在被分析人群中,存在人群迁徙、地理差异、饮食限制等混杂因素,这些因素可能引入与低氧无关的干扰信号。
在一项针对3000余名藏族人群和7000余名东亚血统非藏族人群的大规模全基因组关联研究中,研究人员在9个基因组位点检测到低氧适应信号,其中一个位点对应于EPAS1基因[287]。另一项针对高海拔人群的全基因组关联研究发现,氧气感知机制是介导适应性表型调整的关键因素[288]。EGLN1和EPAS1基因在决定高海拔低气压低氧环境下应答差异方面发挥重要作用,且与红细胞增多症及相关异常的发生存在关联[289]。
总体而言,当基因表达对低氧刺激过度应答时,似乎会出现适应不良模式。例如,氧气感知系统的过度激活会导致缺氧诱导因子(HIF)过表达,进而引发红细胞生成过多及慢性高原病(CMS)相关病理改变。值得注意的是,在暴露于慢性低氧环境(动脉血氧分压=90 mmHg,相当于海拔4630米,持续4周)的北美鹿鼠中,抑制HIF-2α可减轻颈动脉体增生和球细胞增殖,且对血液学指标无影响[255]。
5.2 代谢组学与氨基酸
氨基酸不仅是合成蛋白质的基本单位,还可能作为信号分子发挥重要作用。尽管目前关于低氧与氨基酸相关性的研究较为有限,但氨基酸很可能深度参与低氧应答乃至低氧适应过程。毫无疑问,借助组学技术,未来我们有望更深入地探究氨基酸在低氧适应中可能发挥的作用。
一项针对健康低海拔人群的代谢组学研究显示,当受试者在海拔3800米/氧气浓度13.2%的环境中暴露长达10个月时,其体内主要脂质水平略有下降,而氨基酸代谢则发生显著改变,具体表现为血浆中精氨酸、谷氨酰胺、苯丙氨酸、色氨酸和酪氨酸水平升高[140]。在急性低氧环境(暴露19天,最高海拔6885米/氧气浓度8.8%)中,受试者血浆中色氨酸和血清素水平也出现了类似升高[290]。
大量研究表明,当人类暴露于长期生理性或病理性低氧环境时,血浆中部分(而非全部)氨基酸水平会升高。这一现象可能与低氧相关的蛋白质分解代谢有关[291],但由于低氧既未改变蛋白质ogenic氨基酸库的总量,也未使其随时间发生变化[140],这提示低氧诱导的蛋白质分解代谢并不充分。因此,上述血浆氨基酸水平的变化表明,存在某些以氨基酸为信号分子(而非蛋白质合成原料)的通路。
在涉及氨基酸的通路中,谷氨酰胺/谷氨酸平衡具有重要意义,因其参与脑认知功能调节和通气频率调控。事实上,谷氨酸水平降低会刺激通气活动,以补偿血氧饱和度的下降[292];而谷氨酰胺水平过高则可能通过破坏氨与谷氨酸的平衡,影响脑认知功能[293]。这种平衡紊乱(伴随N-乙酰腐胺、精胺和鸟氨酸水平降低)还会影响血管生成和生殖生理功能[294]。
血浆色氨酸水平升高可能反映色氨酸羟化酶活性受损——该酶需氧气参与,且是血清素合成的关键限速酶[295]。脑内血清素水平下调会导致抑郁、食欲减退和睡眠模式紊乱,这些症状在高海拔居民中较为常见[296,297]。
在急性呼吸窘迫综合征(ARDS)[298]、哮喘[299]等病理性低氧状态下,常可观察到谷氨酸、苏氨酸、牛磺酸、赖氨酸、精氨酸和脯氨酸的代谢异常。在实验性围产期窒息和新生儿缺氧缺血性脑病中,与氨基酸相关的通路也会出现上调[300];而在胎儿生长受限的妊娠案例中,犬尿氨酸通路会被激活[301]。该通路之所以重要,是因为与其相关的烟酰胺通路会促进肺血管重构[302]——这是慢性低氧环境下的常见表现[303]。目前,烟酰胺[304]和烟酸[305]正被作为潜在药物,用于研究其改善低氧诱导性肺血管重构进展的效果。
5.3 研究局限性
本研究未探讨某些尚未明确体现适应过程的生物系统相关问题,例如免疫系统和微生物组。将肿瘤组织视为病理性低氧应答者的理论依据在于:肿瘤可被视为低氧适应成功的范例——相较于健康细胞,肿瘤细胞在低氧环境下的存活能力和增殖能力更强。然而,本研究未大量采用这一理论,因为该可能性尚未得到充分证实,且可能带有推测性。
牦牛[306,307]、美洲驼[308]、鼠兔[309]等多种地理隔离的高海拔哺乳动物,均被视为低氧进化适应的研究模型。利用这些模型开展比较研究,确实可为验证人类低氧适应模式的可靠性和适用性提供重要实验基础。但需注意的是,每种动物均具有物种特异性特征,因此需针对不同动物种类和不同遗传性状分别进行分析,才能与人类的低氧适应模式进行有意义的比较。
尽管明确这一局限性,本综述仍选择聚焦于人类群体数据以及受控的体外和体内实验室研究结果,而未纳入高海拔动物相关的有价值研究(尤其是组学研究)。此外,由于篇幅限制,本研究未能涵盖许多重要研究成果,对此我们深表遗憾。
6. 结论
在地球演化史上,大气氧气浓度曾在0%-30%的范围内波动[310]。尽管各类生物曾多次成功适应不同的氧气水平,但人类是否已丧失适应低于正常氧气浓度环境的能力,目前仍不明确。分析氧气从大气到线粒体传递链中的“瓶颈”,或许能帮助我们确定人体在低氧适应中成功或失败的关键环节。值得注意的是,当前分析的许多环节与70年前一篇文章中预测的结果高度吻合[311]。
研究靶点、分析方法的巨大差异,以及被分析个体或人群中存在的干扰因素,在很大程度上可能模糊了对“氧气传递链各环节应答如何协同促进低氧适应”的判断。部分应答似乎与渐进式适应相符,例如氧气感知机制、缺氧诱导因子(HIFs)、抗氧化防御系统构建、能量代谢调整及一氧化氮(NO)介导的应答等。
与之相反,颈动脉体(CB)对氧气变化的过度敏感(伴随过度通气)、不受控的红细胞生成以及铁代谢异常,可能是适应不良的标志。而心输出量变化、血红蛋白-氧气亲和力改变等其他应答,在低氧适应中似乎未发挥重要作用。
低氧适应似乎需要在持续低氧环境中历经数千年才能完成,这一现象表明人类并非特别适合在高海拔环境中生存。正如已有研究指出的[312],这一结论应促使高海拔研究方向发生转变:当前研究多以来自西方、受过教育、工业化程度高且富裕国家(WEIRD国家)的低海拔人群为研究对象,探索其登高后的生理变化;未来研究应更多聚焦于高海拔常住人群,深入解析其基因、细胞及分子生理学层面的适应机制。
转载本文请联系原作者获取授权,同时请注明本文来自孙学军科学网博客。
链接地址:https://wap.sciencenet.cn/blog-41174-1499668.html?mobile=1
收藏