
 精选
精选
衰老通过重新编程铁稳态限制干细胞特性和肿瘤发生
摘要
衰老与成体干细胞数量和适应性的下降有关。1,2 衰老相关的干细胞特性丧失被认为会抑制肿瘤发生3,4,但这一假设尚未在体内得到验证。在此,我们利用生理性老化的自体基因工程小鼠模型5,6和原代细胞5,6来证明,衰老通过降解肺泡起源细胞的干细胞特性来抑制肺癌的起始和进展。这种表型的基础是小鼠和人类起源细胞中衰老相关转录因子NUPR1及其下游靶标脂联素-2的诱导,导致衰老细胞功能性铁不足。遗传失活NUPR1-脂联素-2轴或补充铁可以恢复干细胞特性并促进老年肺泡细胞的肿瘤生成潜力。相反,针对NUPR1-脂联素-2轴对年轻的肺泡细胞不利,因为它会诱导铁死亡。衰老相关的特定增强子位点的DNA低甲基化与NUPR1表达增加有关,这可以通过抑制年轻肺泡细胞中的DNA甲基化来重现。我们发现,衰老驱动了功能性铁不足,导致干细胞特性丧失和肿瘤发生减少,但促进了对抗铁死亡的抵抗力。这些发现对于再生医学和癌症预防中细胞铁稳态的治疗调节具有重要意义。此外,我们的发现与大多数人类癌症在年轻时发起的模式一致,从而强调了将癌症预防努力指向年轻人的重要性。
正文
癌症可以在人类的所有年龄段发生,但其发病率在第六和第七个十年急剧上升3,7,8,9,10。这种增加与成人干细胞和祖细胞中体细胞突变的积累有关3,9,这些细胞是大多数癌症类型的起源细胞8。然而,随着年龄的增长,干细胞和祖细胞的数量和适应性也会下降1,2,11,12,这被认为可以抵消体细胞突变的积累并抑制肿瘤发生3,4。衰老相关的干细胞适应性损失如何影响肿瘤发生尚未得到调查。
端粒缩短、基因组不稳定、代谢压力、细胞衰老、细胞间通讯失调和表观遗传改变是“衰老的标志”,它们在组织中具有多种有害效应,其中之一就是降解干细胞特性,即干细胞自我更新和分化的潜能13,14。DNA甲基化模式的典型变化,包括基因增强子的低甲基化,是跨物种和细胞类型最普遍的分子生物标志物14,15,16,17,18,19。衰老相关的DNA甲基化变化是否会影响上皮干细胞(大多数人类癌症的起源细胞)的肿瘤生成潜力尚不清楚。
人类衰老相关的改变,包括DNA甲基化的变化,在很大程度上在小鼠中得到了保留,这使得小鼠成为研究哺乳动物衰老的主要模型生物14。小鼠在2岁后出现衰老相关的表型和分子变化,相当于人类的65-70岁14。使用将同种异体癌细胞系移植到年老和年轻宿主小鼠中的研究表明,我们对年老宿主肿瘤微环境变化的理解有所提高,这些变化对肿瘤进展有实质性影响20。尽管这对于模拟晚期肿瘤很有用,但基于移植的方法并不理想地适用于研究衰老如何影响肿瘤发生的早期阶段。特别是,关于衰老相关的细胞内在变化如何影响肿瘤启动和进展的研究很少。
肺腺癌(LUAD)是最常见的肺癌亚型,约占全球所有癌症死亡率的7%7。LUAD的中位诊断年龄为71岁,衰老与吸烟者和非吸烟者中LUAD发病率的增加相关21。然而,这种发病率在80-85岁之后开始下降7。了解衰老如何影响LUAD生物学具有临床重要性,尤其是因为年轻和非常年老的患者由于病例稀少7,22以及因合并症和虚弱被排除在外13,14,而在临床试验中代表性不足。LUAD主要起源于肺泡II型细胞(AT2)5,6,23,这些细胞通过自我更新和在生理性转换及损伤后替换肺泡I型细胞(AT1),充当肺泡的临时干细胞24。
从基因工程小鼠模型中获得的关于LUAD生物学和治疗的重要见解。在最常用的模型中,AT2细胞中的病毒表达Cre重组酶导致致癌性KRAS(G12D)的体细胞激活和p53肿瘤抑制因子的缺失(Kraslox-stop-lox(LSL)-G12DTrp53flox/flox(KP)模型)5,6。KP肿瘤由单个AT2细胞从头发生,并重现了人类LUAD进化的关键分子和组织病理学特征5,6,23。在这里,我们利用KP模型通过在体内启动一个明确的生理老化起源细胞中的强效致癌突变来研究LUAD的发展。
衰老降低AT2细胞的肿瘤生成能力
为了研究衰老对肺癌发展的影响,我们在年轻(12-16周龄)和年老(104-130周龄)的KP小鼠中使用编码Cre重组酶的腺病毒载体,该载体置于AT2特异性表面活性蛋白C启动子的控制下25(AdSPC-Cre;图1a)。年老小鼠的中位生存期比年轻小鼠增加了37%(扩展数据图1a)。为了探究这一生存期增加的原因,我们在LUAD进展的不同阶段检查了KP小鼠肺部的肿瘤负担:非典型腺瘤性增生(AAH;肿瘤启动后4周);腺瘤(肿瘤启动后8周);腺瘤向腺癌过渡(肿瘤启动后12周);以及完全形成的腺癌(肿瘤启动后17周)(图1a)。与年轻的对照组相比,我们在年老小鼠的所有时间点都观察到肿瘤数量大幅减少(图1b、c和扩展数据图1b),这表明衰老抑制了肿瘤启动。支持这一结果的是,携带Rosa26LSL-tdTomato-Cre重组报告子的年老KP小鼠在肿瘤启动后2周显示出与多细胞新生物结节相比,单tdTomato+细胞的比例显著增加(扩展数据图1c)。这一结果表明,年老的Kras突变AT2细胞具有较低的肿瘤生成能力。值得注意的是,AdSPC-Cre载体转导的AT2细胞绝对数量在年老和年轻的Rosa26LSL-tdTomato-Cre报告子小鼠之间没有差异(扩展数据图1d)。对KP小鼠在其生命周期内体内的AT2细胞分析显示,随着年龄的增长,AT2细胞的肿瘤生成潜力显著下降(扩展数据图1e)。
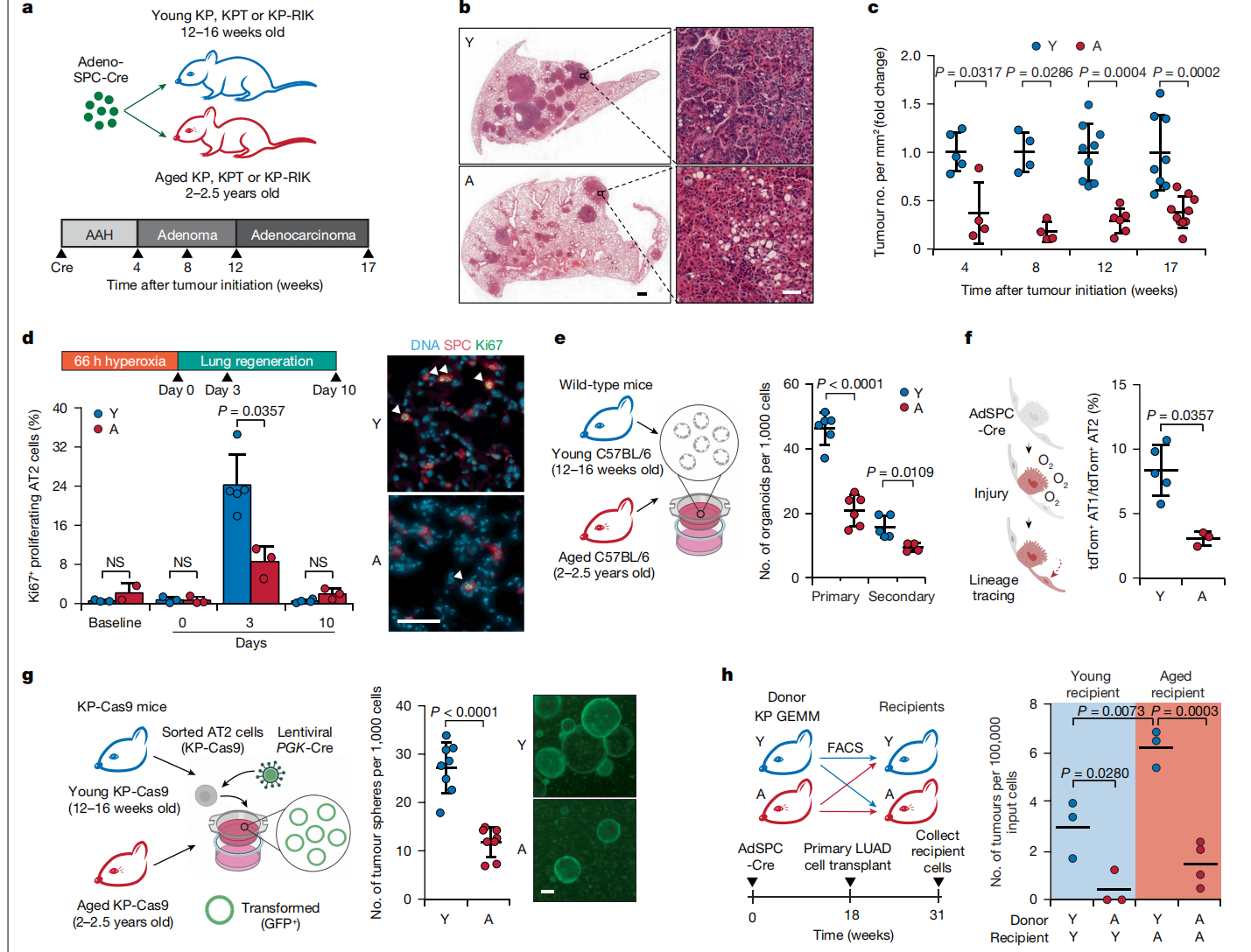
图1:年老的AT2细胞表现出再生和肿瘤生成的内在潜力降低。
a,本研究的实验方案。KP-RIK KrasLSL-G12D/+Trp53fl/flRosa26LSL-rtTA-IRES-mKate2/+小鼠。b,肿瘤(腺癌)启动后17周10只年老(A)和8只年轻(Y)KP小鼠的肺代表图像。比例尺 1毫米(左)和50微米(右)。c,不同时间点肿瘤启动后年老和年轻KP小鼠肿瘤负担的变化倍数(n(从左到右)=5, 4, 9和8只年轻小鼠以及4, 4, 6和10只年老小鼠)。d,基线(稳态)和高氧损伤后第0天、3天和10天肺泡再生期间AT2细胞增殖的定量(左)和图像(右)(n(从左到右)=3, 3, 5和4只年轻小鼠以及2, 3, 3和3只年老小鼠)。箭头指示增殖的AT2细胞。比例尺 20微米。e,来自年老和年轻初级AT2细胞的肺泡类器官形成测定的示意图(左)和定量(右)(n = 6个生物重复用于初级培养和n = 5和4个生物重复用于次级培养)。f,用tdTomato标记的年老和年轻AT2细胞谱系追踪的示意图(左)和定量(右)(n = 5只年轻和3只年老小鼠)。tdTomato+ AT1细胞的百分比(AT1标志物podoplanin+tdTomato+)相对于tdTomato+ AT2细胞的数量(SPC+tdTomato+)。g,使用从KP-Cas9小鼠分离的初级AT2细胞进行的离体转化测定的示意图(左)、定量(中)和图像(右)(n = 8个生物重复)。PGK,磷甘油酸激酶1启动子。比例尺 200微米。h,异时移植KP肿瘤细胞的示意图(左)和定量(右)(n(从左到右)=3, 3, 3和4只小鼠)。GEMM,基因工程小鼠模型。数据以平均值±标准差表示(c–h)。P值使用双尾曼-惠特尼检验(c,d,f)、双尾学生t检验(e,g)或单向方差分析(ANOVA)(h)计算。NS = 不显著。e–g中的插图使用BioRender创建(https://biorender.com)。
为了测试这些发现是否在KP基因型之外也具有相关性,我们利用CRISPR介导的Eml4–Alk融合事件26(扩展数据图1f)在年老和年轻野生型小鼠中启动了肺部肿瘤,这代表了另一种临床上相关的LUAD亚型。与年轻小鼠相比,年老小鼠在肿瘤数量和整体肿瘤负担上显示出显著减少(扩展数据图1g,h),尽管年老和年轻小鼠的AT2细胞在体内发生Eml4–Alk融合事件的频率相似(扩展数据图1i)。因此,肺上皮中的肿瘤发生潜力的丧失是独立于驱动癌基因或p53状态的。
肿瘤起始潜力与组织中起源细胞的自我更新能力和分化能力(干细胞特性)密切相关8。与年轻小鼠的肺相比,我们发现年老(年老AT2细胞)野生型小鼠的AT2细胞密度减少了47%(扩展数据图1j)。此外,衰老导致AT2细胞在总肺上皮细胞池中的比例逐渐减少(扩展数据图1k,l),这表明随着年龄的增长,AT2细胞的数量比其他肺上皮细胞下降得更多。为了探究体内AT2细胞的自我更新能力,我们通过对年老和年轻野生型小鼠进行高氧损伤27来刺激肺泡再生。与年轻小鼠相比,年老小鼠在损伤后3天(肺泡再生的高峰增殖期)增殖(Ki67+)AT2细胞的比例减少了69%(图1d)。为了研究体外环境中受控且一致条件下AT2细胞的内在自我更新能力,我们对分离的初级AT2细胞进行了3D肺泡类器官测定28。我们发现,与年轻小鼠相比,从年老小鼠中分离的AT2细胞形成的类器官数量减少了54%,这一结果与已发表的数据一致29。类器官生长能力的降低在次代培养中持续存在(图1e和扩展数据图1m),这一结果进一步支持了年老AT2细胞自我更新能力下降是由于细胞内在变化的观点。值得注意的是,年老和年轻小鼠的AT2细胞中几乎不存在与衰老相关的β-半乳糖苷酶活性,尽管在年老小鼠的肺部观察到非上皮衰老细胞的增加(扩展数据图1n)。
接下来,我们测试了年老AT2细胞恢复肺泡完整性的能力,这是依赖于AT2细胞的肺再生过程24。为此,我们通过测量肺泡空间的平均弦长(MCL)来评估损伤后的肺泡气腔。值得注意的是,即使在没有损伤的情况下,年老小鼠的肺泡气腔已经扩大,这与随着年龄增长AT2细胞再生能力累积性损失的结果一致。这些肺泡在损伤后4周进一步扩大,并且与基线相比仍然显著增大,而年轻小鼠的肺泡则恢复到基线水平(扩展数据图2a)。接下来,我们在年老和年轻Rosa26LSL-tdTomato小鼠中追踪了AT2细胞,然后进行了高氧损伤。与年轻AT2细胞相比,年老AT2细胞分化为AT1细胞的能力大幅下降(图1f)。综合这些结果表明,衰老抑制了AT2细胞的干细胞特性。
为了直接探究转化能力,我们从年老和年轻的KrasLSL-G12D/+Trp53fl/flRosa26LSL-Cas9-2a-eGFP/+(KP-Cas9)小鼠中分离出初级AT2细胞,并通过慢病毒递送Cre在体外肿瘤球形成测定中诱导转化。与体内实验类似,年老AT2细胞的转化效率显著受损(图1g),即使调整了年老和年轻AT2细胞之间肺泡类器官形成的基线差异也是如此(扩展数据图2b)。这种差异并非由于慢病毒转导效率的差异,无论是在体外还是体内,年老和年轻AT2细胞的慢病毒转导效率都是相似的(扩展数据图2c,d)。值得注意的是,AT2细胞自我更新能力(图1d,e)和分化能力(图1f)的50–70%的减少与年老AT2细胞整体约60%的肿瘤发生潜力减少紧密相关(扩展数据图1c)。这一结果表明,AT2细胞干细胞特性的丧失导致年老肺部肿瘤发生潜力的降低。即使在移除了年老组织微环境的体外条件下,年老AT2细胞仍显示出生长和转化潜力的缺陷。这一发现表明,衰老通过细胞内在机制抑制了AT2细胞的干细胞特性和肿瘤发生。
为了直接测试肺部微环境的贡献,我们从年老小鼠和年轻供体小鼠中分离出初级自体KP LUAD细胞,并将它们异时移植到年老和年轻的同基因受体小鼠的肺部。无论受体的年龄如何,来自年轻供体的KP细胞形成肿瘤的效率显著高于来自年老供体的细胞。此外,来自年轻供体的KP细胞在年老受体中形成了更多的肿瘤(图1h)。这些结果表明,年老的肺部微环境可能更有利于LUAD肿瘤的发生。然而,尽管有这种效应,年老小鼠仍然发展出较少的肿瘤,这是由于AT2细胞转化能力的固有损失所致。
衰老延缓肺部肿瘤进展
除了启动频率降低外,与年轻对照组相比,老年KP小鼠形成的肿瘤在4、8和17周的肿瘤发育过程中体积减少了40-60%;然而,在12周时没有观察到差异(扩展数据图2e)。KP肿瘤的增殖指数在所有时间点上老年小鼠显著较低(扩展数据图2f)。相反,我们在老年肿瘤与年轻肿瘤中未检测到衰老细胞数量的差异(扩展数据图2g-i)或切割caspase-3+凋亡细胞的差异(扩展数据图2j)。这些结果表明,较小的肿瘤体积是由于增殖减少而不是衰老或凋亡增加所致。
除了增殖减少外,老年KP小鼠的肺部在第12周显示出较高比例的1级AAH病变,以及在肿瘤启动后的第17周显示出较高比例的2级腺瘤,相比之下年轻KP小鼠则较少(图1b及扩展数据图2k,l)。相反,年轻小鼠在第17周显示出比老年小鼠更高比例的4级晚期腺癌(扩展数据图2l)。这些发现表明,老年KP小鼠的肿瘤在组织学上进展缓慢。为了研究老年和年轻小鼠中LUAD肿瘤的分子演变,我们使用单细胞mRNA测序(scRNA-seq)对肿瘤启动后4、12和17周的KP LUAD细胞进行了分析。同时收集了野生型老年小鼠和年轻小鼠的AT2细胞进行分析。无监督聚类scRNA-seq数据揭示了六个具有不同表达模式的簇。其中五个对应于我们在LUAD演变过程中先前识别的癌细胞状态,而野生型AT2细胞形成了它们自己的簇(扩展数据图3a)。尽管老年和年轻小鼠的肿瘤沿着相似的轨迹进展,但老年小鼠的肿瘤显示出显著的延迟进展(扩展数据图3b-d)。例如,在4周时,年轻小鼠的KP细胞比老年小鼠更多进展到了高塑性细胞状态——早期进展的一个指标(扩展数据图3b,c)。在17周时,与老年小鼠的KP细胞相比,更多年轻小鼠的KP细胞处于内胚层样状态(扩展数据图3d)——这是进展晚期发生的转变。通过免疫染色验证了我们的scRNA-seq发现,分别在早期和晚期进展时间点标记高塑性状态(integrin-α2和keratin-8)和内胚层样状态(HNF4α)(扩展数据图3b-d)。
接下来,我们利用体外AT2细胞转化实验(图1g)模拟肿瘤演变,并通过bulk RNA-seq在八个传代过程中(约3个月)分析了生成的肿瘤球体。新鲜分离的初级AT2细胞和来自老年和年轻野生型小鼠的初级AT2类器官培养物作为基线对照进行分析(图1e,g和扩展数据图3e)。尽管体外KP肿瘤球体的转录组特征与体内LUAD肿瘤不同(补充表1),但从老年AT2细胞启动的肿瘤球体在无监督扩散伪时间轨迹上的进展显著延迟(扩展数据图3f,g)。这种延迟体现在从老年AT2细胞衍生的肿瘤球体中肺泡标志物的下调速度较慢(扩展数据图3h)。这些发现表明,由衰老引起的AT2细胞内在变化延缓了LUAD肿瘤的进展。
Nupr1抑制老年肺中的肿瘤发生
为了揭示AT2细胞转化潜力丧失背后的分子机制,我们对包含老年和年轻AT2细胞及LUAD细胞的scRNA-seq数据进行了差异基因表达分析。我们在AT2细胞和LUAD细胞之间检测到强烈的年龄依赖性相关差异表达基因(DEGs)(图2a)。基于两部分广义统计模型(方法),170个基因——占老年和年轻AT2细胞中DEGs的8.2%——在AT2细胞中和所有5种转录细胞状态中显示出一致的表达变化,这些细胞状态分子定义了LUAD进展(扩展数据图3i和补充表2)。这些结果表明,一部分与衰老相关的基因表达变化在AT2细胞起源和从中产生的LUAD肿瘤之间共享,并且这些变化在肿瘤进展的不同癌细胞状态中持续存在。
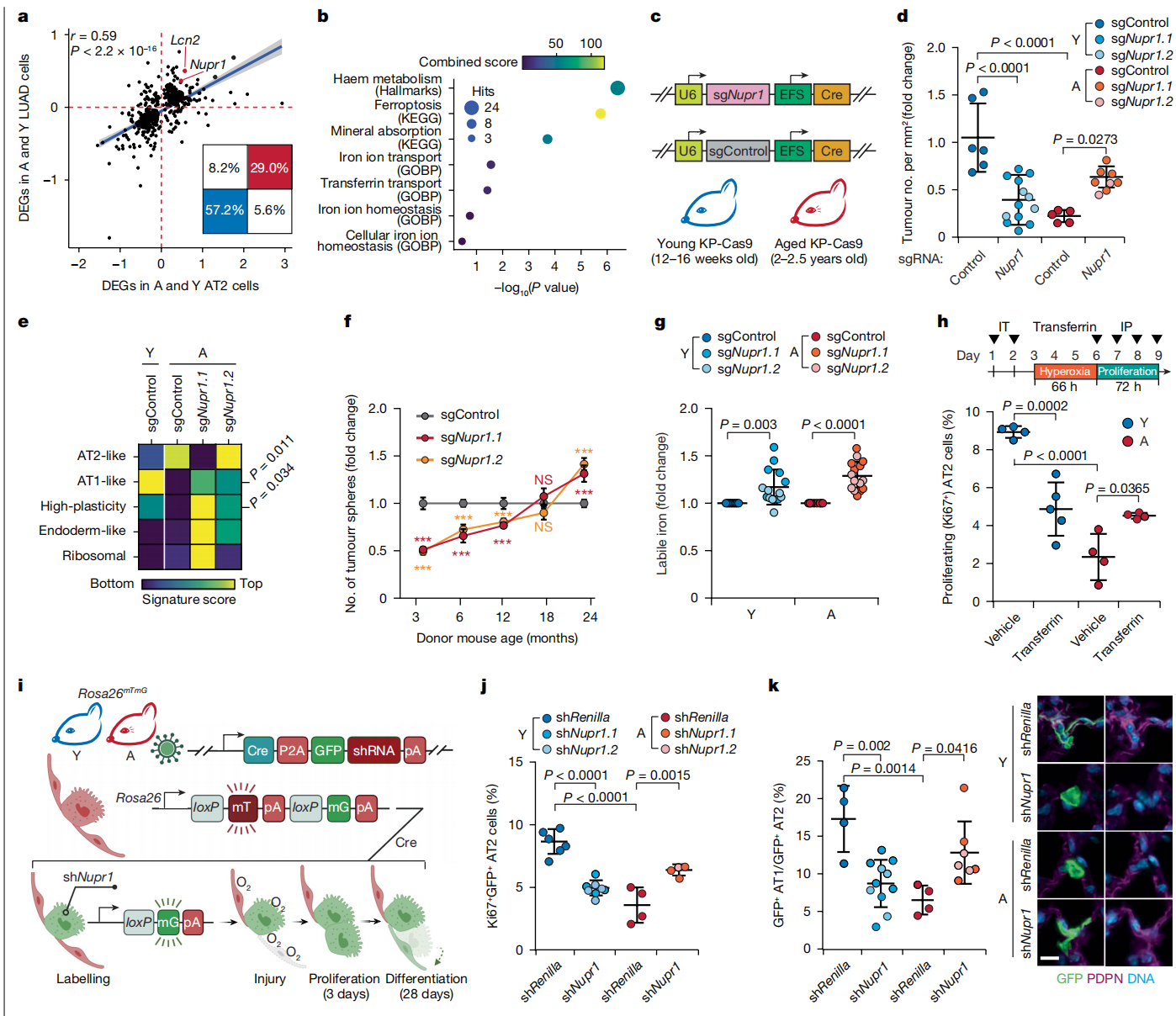
图2:诱导Nupr1通过破坏铁稳态抑制老年肺中的肿瘤发生
a,老年和年轻正常AT2细胞(x轴)与LUAD细胞(y轴)之间的DEGs的皮尔逊相关系数。插图显示每个象限中的基因百分比(二项检验)。回归线(蓝色)及其95%置信区间(灰色)被绘制出来。b,老年AT2和LUAD细胞签名中富集的铁代谢相关基因集(在补充表2中定义)。c,体内LUAD肿瘤发生的慢病毒Nupr1基因靶向策略示意图。d,c中生成的小鼠肿瘤负担(n(左至右)= 6 13, 5和8只小鼠)。e,从c中获得的KP-Cas9细胞的bulk RNA-seq显示富集了KP LUAD细胞状态签名(在扩展数据图3a中定义),这些签名定义了进展。f,3-24月龄KP-Cas9小鼠AT2细胞的体外转化,用sgControl或sgNupr1转导。肿瘤球体数量以sgControl为基准进行标准化(n = 4次技术重复)。数值P值见补充表18。g,Nupr1敲除后体外转化的LUAD细胞中不稳定铁的变化倍数。FerroFarRed染色的平均荧光强度以sgControl为基准进行标准化(n(左至右)= 9, 15, 8和15个生物重复)。h,给予载体或转铁蛋白的年轻和老年C57BL/6小鼠中AT2细胞增殖的方案(顶部)和量化(底部)(IT,气管内插管;IP,腹腔注射)(n(左至右)= 4, 5, 4和4只小鼠)。i-k,评估Nupr1敲减后AT2细胞增殖(j;n(左至右)= 6, 8, 4和4只小鼠)和分化(k;n(左至右)= 4, 11, 4和7只小鼠)的实验策略(i),并在损伤后3天(j)或28天(k)进行评估。PDPN是AT1细胞标记。表达shRNA的细胞通过GFP荧光可视化。标尺= 10μm。数据表示均值±标准差(d, f-h, j,k)。P值使用双尾学生t检验(f,g)或单因素方差分析(d, h-k)计算。插图i使用BioRender创建(https://biorender.com)。
对AT2细胞和LUAD细胞中由衰老改变的基因程序进行基因集富集分析,一致显示涉及铁稳态的基因被诱导(图2b,扩展数据图4a和补充表3)。在这些途径中被衰老最显著诱导的基因是核蛋白转录调控因子1(Nupr1)(扩展数据图3i和4b),这是一种应激诱导的转录共激活因子,控制铁稳态32,33。除了铁代谢外,NUPR1还调节一系列其他细胞过程,包括DNA修复、内质网应激、氧化应激反应、细胞周期、自噬、凋亡和染色质重塑33,34。然而,在我们的数据集中这些其他途径的变化不太明显或在不同进展阶段不一致(补充表3)。
我们证实,在老年野生型小鼠的AT2细胞和老年小鼠的LUAD肿瘤中,Nupr1 mRNA被原位诱导(扩展数据图4c,d)。接下来,我们在老年小鼠和年轻KP-Cas9小鼠的肺部通过慢病毒共投递单向导RNA(sgRNA)和Cre,在体内启动时失活Nupr135(图2c)。与已发表文献将NUPR1识别为抗癌靶标33,34一致,失活Nupr1在肿瘤启动后的12周抑制了年轻小鼠中的肿瘤发生。然而,针对老年AT2细胞的Nupr1靶向促进了体内外的肿瘤启动(图2d和扩展数据图4e)。同样,给予NUPR1核输入的小分子抑制剂ZZW-115(参考文献36)增加了来自老年小鼠的KP AT2细胞的体外肿瘤球形成。相比之下,抑制年轻小鼠KP AT2细胞的球形成(扩展数据图4f)。此外,针对老年小鼠的Nupr1靶向导致肺肿瘤平均大小增加(扩展数据图4g)。老年小鼠肿瘤中失去Nupr1使癌细胞增殖增加到与年轻小鼠对照肿瘤相似的水平,而年轻KP癌细胞则观察到相反的情况——增殖受到抑制(扩展数据图4h)。RNA-seq分析表明,Nupr1的缺失导致老年小鼠KP肺肿瘤进展到比老年小鼠对照肿瘤在此时间点观察到的年轻小鼠晚期分子阶段。这表明Nupr1作为老年小鼠肿瘤进展的屏障(图2e和补充表4)。遗传失活Nupr1强烈抑制了从3个月大小鼠分离的AT2细胞的转化。然而,随着年龄增长,与对照sgRNA相比,Nupr1敲除的效果逐渐减弱,在18个月时变得中性,直到24个月时最终促进肿瘤发生(图2f)。综上所述,这些结果表明,与衰老相关的NUPR1表达诱导通过细胞自主机制抑制了AT2细胞转化。
增加的NUPR1破坏铁稳态
用去铁胺(DFO)螯合铁削弱了老年小鼠KP-Cas9 AT2细胞在遗传或药理学靶向NUPR1情况下肿瘤球形成的增加(扩展数据图4i,j)。相反,DFO在年轻小鼠背景下NUPR1失活的情况下对肿瘤球没有影响(扩展数据图4i,j)。这些发现表明,与衰老相关的Nupr1表达诱导导致AT2细胞功能性铁缺乏。与这一概念一致的是,补充老年AT2细胞以转铁蛋白结合的铁或柠檬酸铁铵(FAC)在体外转化试验中促进了肿瘤球形成(扩展数据图4k,l)。此外,添加转铁蛋白结合的铁并未改变年轻或老年AT2细胞体外转化能力中Nupr1敲除的效果(扩展数据图4m)。为了测试铁剥夺的效果,我们在存在铁捕获剂2,5-二羟基苯甲酸(2,5-DHBA)的情况下进行了体外转化。2,5-DHBA降低了年轻和老年细胞中的可变铁水平(扩展数据图5a)。2,5-DHBA进一步抑制了老年体外转化AT2细胞的生长,而年轻转化AT2细胞的生长则因2,5-DHBA补充而增加(扩展数据图5b)。这些结果表明,AT2细胞在衰老过程中发展出功能性铁充足,而在年轻细胞中铁是生长限制因素。
为了直接测试NUPR1是否控制转化AT2细胞中的铁水平,我们测量了可变细胞铁的水平。失去Nupr1促进了从老年和年轻小鼠分离的转化AT2细胞的铁摄取(图2g和扩展数据图5c,d)。值得注意的是,可变和总铁的基础水平在老年AT2细胞中高于年轻细胞(扩展数据图5e,f),这表明老年AT2细胞对铁的需求增加,铁的错误定位或储存铁的动员效率低下。
我们接下来研究了由增加的Nupr1表达引起的功能性铁不足是否是老年AT2细胞失去干细胞性和再生能力的原因。确认我们的体外结果,转铁蛋白结合的铁或靶向Nupr1的短发夹RNA(shNupr1)部分逆转了老年小鼠体内高氧损伤后AT2细胞自我更新能力的降低,但对年轻AT2细胞的自我更新有害(图2h–j和扩展数据图5g)。在培养的AT2类器官中补充转铁蛋白或化学抑制NUPR1核输入后得到了类似的结果(扩展数据图5h,i)。为了研究细胞的分化潜力,我们通过对老年和年轻Rosa26mTmG/+小鼠进行气管介导的慢病毒shRNA靶向Nupr1或Renilla荧光素酶(对照)和Cre,随后进行高氧损伤,对AT2细胞进行谱系追踪(图2i)。在这个实验中,携带shRNA的谱系追踪AT2细胞可以通过GFP表达来追踪。敲低Nupr1逆转了老年小鼠中AT1分化的损失。然而,年轻AT2细胞向AT1谱系的分化因Nupr1失活而减少(图2k)。因此,由Nupr1诱导引起的与老化相关的功能性铁不足抑制了AT2细胞的干细胞性。
NUPR1通过脂笼蛋白-2破坏铁稳态
NUPR1通过诱导铁隔离蛋白脂笼蛋白-2(由Lcn2基因编码)的表达,保护胰腺癌细胞免受过量铁的影响32。在老年AT2细胞和LUAD细胞中,Lcn2被诱导(图2a和3a,b以及扩展数据图5j)。老年LUAD肿瘤中也观察到脂笼蛋白-2的诱导,而Nupr1敲除将其恢复到与年轻小鼠肿瘤相似的水平(图3b)。同样地,对NUPR1进行靶向处理时,老年和年轻AT2细胞在体外转化试验中以及野生型原代AT2细胞体内外都表现出Lcn2的抑制(扩展数据图5k–n)。Lcn2敲除增加了老年AT2细胞的肿瘤球形成能力,并抑制了年轻AT2细胞的转化(图3c,d),基本上重现了Nupr1敲除的表型(图2g和扩展数据图4e)。相比之下,一组其他关键铁运输和储存基因的敲除对年轻和老年AT2细胞在转化试验中同样有害。这一结果表明,NUPR1-脂笼蛋白-2轴具有年龄依赖性功能的独特性(扩展数据图6a)。Lcn2 cDNA完全消除了在敲除Nupr1的细胞中对AT2细胞转化和铁摄取的救援作用(图3e和扩展数据图6b)。这一结果直接表明Lcn2在NUPR1下游的诱导是支撑老化相关功能性铁不足和AT2转化能力丧失的机制。同样,补充重组脂笼蛋白-2减弱了化学抑制NUPR1后老年AT2肿瘤球形成的增加(扩展数据图6c)。相反,用阻断抗体中和细胞外的脂笼蛋白-2增加了来自年轻和老年小鼠细胞中的可变铁(扩展数据图6d),这进而导致老年细胞球体形成的增加和年轻细胞球体生长的抑制(扩展数据图6e)。值得注意的是,Lcn2敲除对生长和铁水平的影响大于阻断抗体的效果。这表明细胞内和细胞外脂笼蛋白-2池对于老年AT2细胞和LUAD细胞中功能性铁不足的发展都很重要。
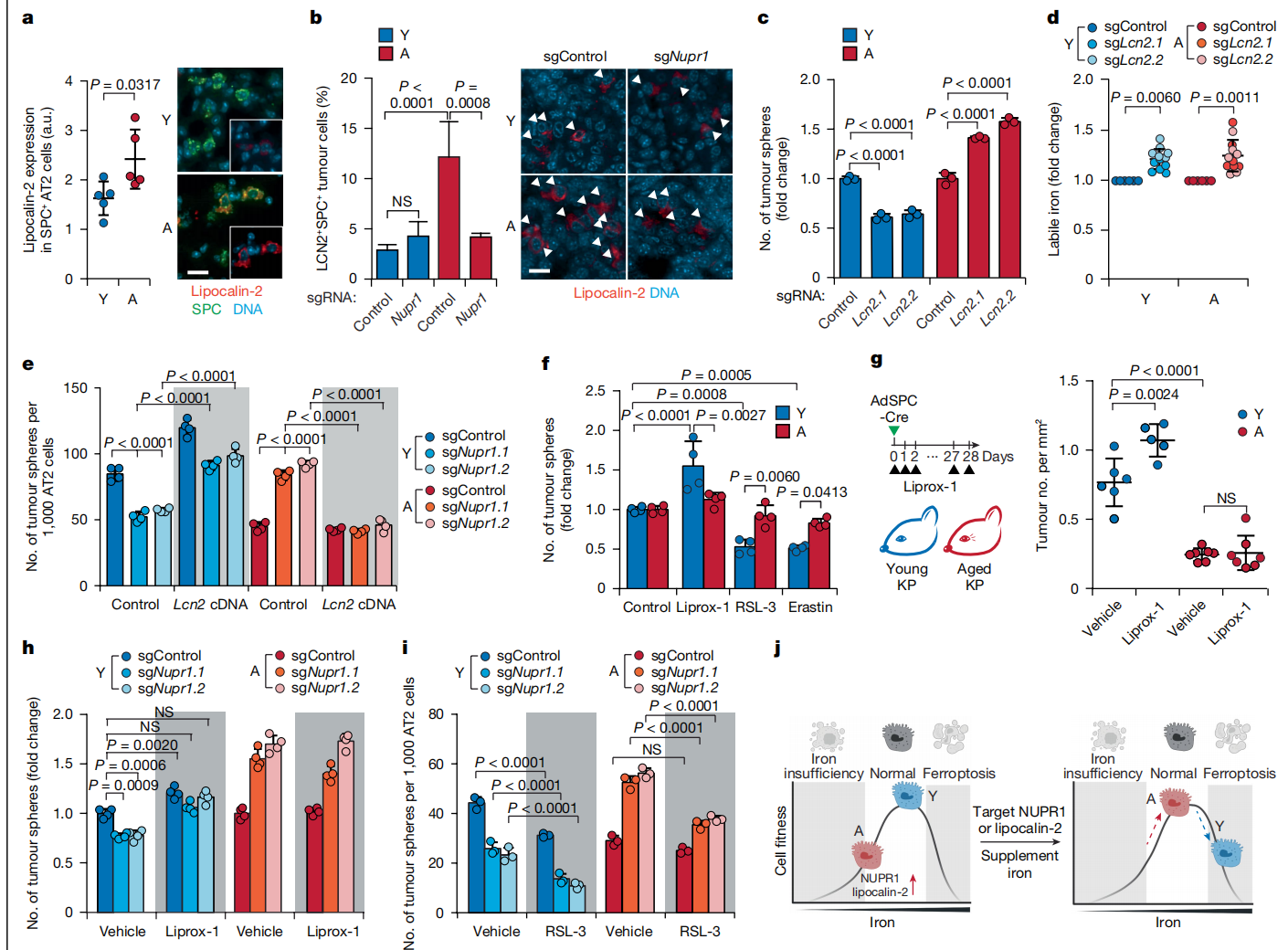
图3:NUPR1通过脂笼蛋白-2驱动老年AT2细胞功能性铁不足和铁死亡抵抗。
a, SPC+ AT2细胞中脂笼蛋白-2的定量(左)和图像(右)(n = 5只年轻和5只老年小鼠)。a.u., 任意单位。b, 肿瘤启动后12周KP-Cas9 LUAD细胞中脂笼蛋白-2+细胞的百分比(左)和图像(右)(n = 31, 30, 10和31个肿瘤)。箭头指示脂笼蛋白-2+ LUAD细胞。为清晰起见省略用于识别LUAD细胞的SPC染色。c, 用对照sgRNA或靶向Lcn2的sgRNA进行的AT2细胞体外转化(n = 3个生物重复)。d, 用sgControl或靶向Lcn2的sgRNA进行的LUAD细胞体外转化中可变铁的变化倍数(n = 6, 12, 6和12个生物重复)。FerroFarRed染色的平均荧光强度归一化至sgControl。e, 用靶向Nupr1的sgRNA,有或没有Lcn2 cDNA进行的AT2细胞体外转化(灰色背景)。N = 4个生物重复。f, 用指定化学品进行的老年和年轻AT2细胞体外转化。肿瘤球数量归一化至对照组(n = 4个生物重复)。Liprox-1即liproxstatin-1。g, 老年和年轻KP小鼠在肿瘤启动后4周的肿瘤数量示意图(左)和定量(右),有或没有liproxstatin-1(n = 6, 5, 7和7只小鼠)。h,i, 用靶向Nupr1的sgRNA,有或没有liproxstatin-1(h; n = 4个生物重复)或RSL-3(i; n = 3个生物重复)进行的AT2细胞体外转化(灰色背景)。j, NUPR1的年龄依赖性功能。数据显示平均值±标准差(a, c–i)或标准误(b)。比例尺20 μm(a, b)。P值使用双尾曼-惠特尼检验(a)或单因素方差分析(b–i)计算。j中的插图使用BioRender (https://biorender.com) 创建。
NUPR1–脂质运载蛋白-2轴的失活对年轻AT2细胞在转化和再生过程中都是有害的。鉴于NUPR1–脂质运载蛋白-2轴已被证明与铁死亡防御有关,我们提出年轻AT2细胞比老年AT2细胞对铁死亡更敏感。通过化学或遗传抑制谷胱甘肽过氧化物酶-4(GPX4)诱导铁死亡,损害了年轻AT2细胞的体外肿瘤球形成能力,而老年AT2细胞则表现出抵抗(图3g和扩展数据图6f, g)。相反,用liproxstatin-1抑制铁死亡促进了年轻AT2细胞的体内外转化,而在老年AT2细胞中未观察到效果(图3g, h)。此外,liproxstatin-1治疗逆转了化学抑制NUPR1对年轻AT2细胞转化的抑制作用(扩展数据图6h)。Nupr1敲除加剧了化学铁死亡诱导剂RSL-3在年轻细胞中的效果,并使老年AT2细胞对RSL-3变得敏感(图3i)。同样,补充结合了铁的转铁蛋白使老年AT2细胞对RSL-3变得敏感(扩展数据图6i)。总之,我们的结果表明,与老化相关的NUPR1–脂质运载蛋白-2轴过度激活导致功能性铁缺乏,这抑制了老年AT2细胞的转化和干细胞特性,同时使其对铁死亡产生抵抗。相反,年轻细胞维持足够的铁水平以促进生长,但对铁死亡敏感(图3j)。
DNA低甲基化诱导NUPR1表达
鉴于DNA甲基化作为老化的主要特征之一的重要性,我们研究了DNA甲基化在AT2细胞和癌细胞共享的老化相关基因表达变化中的作用。我们在年轻和老年AT2细胞以及年龄匹配的LUAD肿瘤中进行了全基因组范围的DNA甲基化评估(扩展数据图7)。与其他类型的细胞一致,我们在老年AT2和LUAD细胞中观察到全局DNA甲基化的丧失,同时伴有CpG岛特异性的高甲基化现象。此外,年龄是无监督样本聚类的最显著驱动因素(扩展数据图7c, d, g, h和补充表5及6)。为了将差异甲基化的胞嘧啶(DMCs)与AT2细胞中的启动子和增强子联系起来,我们将DMCs映射到组蛋白3(H3)修饰(H3K4me1、H3K27ac和H3K4me3)(扩展数据图8a和补充表7)。在老年和年轻样本之间观察到的最显著差异是在由H3K4me1和H3K27ac标记的活动增强子处的高甲基化和低甲基化DMCs的数量上(扩展数据图8a和补充表7)。我们发现,在AT2细胞和LUAD细胞中随年龄变化的共享基因集的启动子和增强子处,DNA低甲基化与基因表达的变化有显著相关性(图2a和4a)。在老年相对于年轻的AT2细胞中,Nupr1在一个由H3K4me3和H3K27ac标记的内含子内以及一个标记为活动增强子的位点(H3K4me1和H3K27ac)处呈现低甲基化,而Lcn2在一个活动增强子位点(H3K4me、H3K4me3和H3K27ac)处去甲基化(扩展数据图8b–d和补充表8)。这些关键基因调控区的DNA甲基化丢失与基因表达随年龄增长而被表观遗传去抑制一致。与AT2细胞类似,AT2和LUAD细胞间老化相关的DNA甲基化变化在构成AT2和LUAD细胞共享的老化相关转录本签名的基因上呈正相关(图4b和补充表2)。
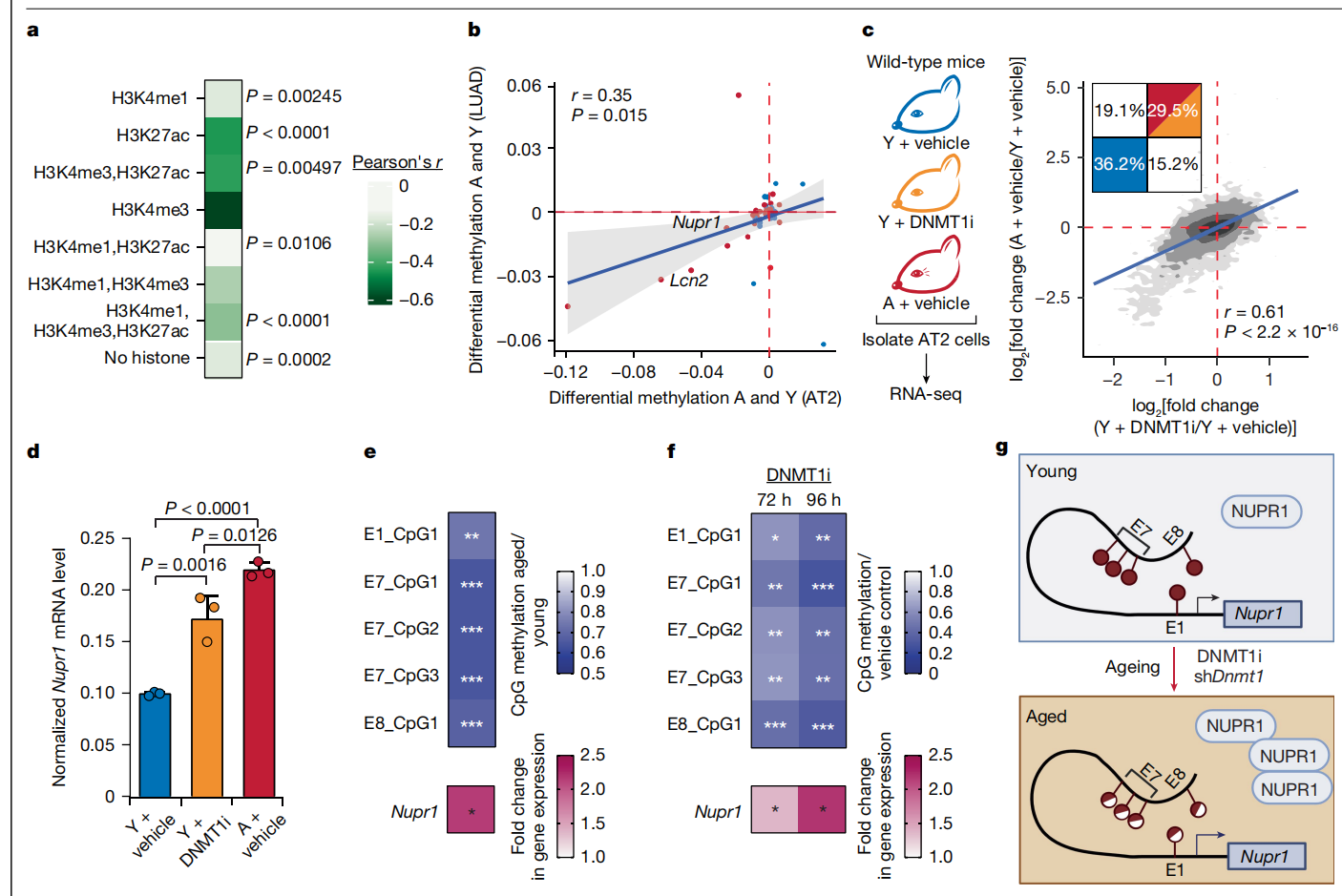
图4:DNA低甲基化支撑老化相关的NUPR1–脂质运载蛋白-2轴的诱导。
a,在每个组蛋白标记类别中,DMCs与DEGs之间的相关性(假发现率(FDR)<0.05)。b,显示在扩展数据图3i中的年龄相关签名基因的启动子区域(TSS上下游1000 bp至+200 bp)的差异甲基化。回归线(蓝色)及其95%置信区间(浅灰色)已绘制。上调基因在老年和年轻AT2及LUAD细胞中分别用红色和蓝色表示。c,方案(左侧)和相关性(右侧),比较年轻AT2细胞在DNMT1i(GSK3484862)处理后体内(x轴)与老年或年轻对照处理小鼠(y轴)的DEGs。灰度区域代表概率分布。回归线以蓝色显示。插图显示每个象限中基因的百分比(二项检验)。d,有无DNMT1i处理的体内转化LUAD细胞中Nupr1的表达(n = 3个生物重复)。e,f,顶部,通过酶促甲基测序测量在原代老年和年轻AT2细胞中定义的Nupr1远端增强子E1、E7和E8处的甲基化变化(e;n = 4只小鼠)以及经DNMT1i处理的培养KP肺癌细胞通过焦磷酸测序测量(f;n = 4个独立细胞系)。结果以相对于年轻AT2细胞(e)或对照处理细胞(f)的倍数变化显示。底部,上述条件下Nupr1表达的热图(n = 7个原代AT2样本(e)和n = 4个KP肺癌细胞系(f))。g,老化相关的低甲基化上调Nupr1。实心红色棒棒糖表示甲基化的CpGs,混合棒棒糖标记低甲基化的CpGs。e和f的数值P值在补充表18中提供。数据显示均值±标准差(d)。相关性或P值使用皮尔逊相关检验(a–c)、单因素方差分析(d)或双尾配对t检验(e, f)计算。g中的插图使用BioRender (https://biorender.com) 创建。
为了功能性地验证DNA低甲基化是否是AT2细胞中与衰老相关的基因表达签名的驱动因素,我们通过全身给药GSK3484862(一种DNA甲基转移酶-1(DNMT1)的小分子抑制剂)在年轻野生型C57BL/6J小鼠中抑制了DNMT1,为期8天(图4c和扩展数据图9a)。接受DNMT1抑制(DNMT1i)的年轻AT2细胞显示出与老年AT2细胞中观察到的转录组变化有很强的相关性。这一结果表明,全局渐进性DNA低甲基化是AT2细胞中衰老转录组的关键机制驱动因素(图4c和补充表9)。在AT2和LUAD细胞之间共享的与衰老相关的基因表达签名中(图2a),Nupr1和Lcn2是由DNMT1i在年轻小鼠的AT2细胞中诱导的前几个基因(图4d,扩展数据图9b,c和补充表9)。值得注意的是,DNMT1i特异性抑制了年轻AT2细胞的转化,而不是老年AT2细胞(扩展数据图9d)。DNMT1i也在此背景下诱导了Nupr1和Lcn2基因表达(扩展数据图9e,f)。为了排除DNMT1i可能的脱靶效应,我们通过慢病毒shRNA(shDnmt1)在Rosa26mTmG/+小鼠体内抑制了Dnmt1(扩展数据图9g–i)。类似于DNMT1i,shDnmt1在年轻AT2细胞中诱导了类似衰老的转录组,包括Nupr1的诱导(补充表10和扩展数据图9j–l)。
为了精确定位低甲基化与老年细胞中Nupr1表达诱导相关的增强子,我们在Nupr1转录起始位点(TSS)±500 kb内确定了11个候选增强子位点。我们根据染色质可及性、位于已知能诱导Nupr1表达的转录因子的结合模序中的衰老相关低甲基化CpG位点以及AT2细胞中活性增强子的组蛋白标记来定义候选增强子(扩展数据图8b–d和10a,b和补充表8)。为了鉴定功能性Nupr1增强子,我们在三个独立的KP LUAD细胞系中进行了阵列化的CRISPR干扰(CRISPRi)筛选(扩展数据图10c和补充表11和12)。这项努力确定了增强子1(E1)(在Nupr1 TSS上游25,846 bp)、E7(在TSS上游113,969 bp)和E8(在TSS上游114,169 bp)为高置信度Nupr1增强子(扩展数据图10d)。高分辨率检查发现E1中的一个CpG、E7中的三个CpG和E8中的一个CpG在AT2细胞中特异地与衰老相关低甲基化(图4e和扩展数据图10e和11a)。值得注意的是,这些CpG在DNMT1i后通过目标特异性焦磷酸测序显示显著低甲基化(扩展数据图11b),这与DNMT1i后Nupr1表达增加相关(图4f,扩展数据图11b和补充表13)。此外,用CRISPRi抑制E1、E7或E8导致Nupr1水平减少约50%(扩展数据图10d),这表明这些增强子强烈调节Nupr1表达。因此,这些增强子与低甲基化相关活性的增加共同可能足以解释我们在老年AT2细胞中观察到的Nupr1表达增加。这些发现符合一个模型,即Nupr1增强子的低甲基化——或许还有启动子和/或内含子的低甲基化的贡献——促进了与衰老相关的Nupr1表达诱导(图4g)。
衰老变化在广泛上是保守的
与我们小鼠中的发现类似,我们在老年人的肺中观察到AT2细胞密度减少(扩展数据图12a和补充表14)。来自老年个体的人AT2细胞显示出NUPR1和脂钙蛋白-2(LCN2)表达增加(图5a和扩展数据图12b)。值得注意的是,用结合铁的转铁蛋白强烈促进了从65-78岁个体分离出的人原代AT2细胞形成的类器官(图5b和补充表15),这表明铁可用性也是老年人类AT2细胞的生长限制因素。接下来,我们分析了来自癌症基因组图谱(The Cancer Genome Atlas)的LUAD整体RNA-seq数据中的与衰老相关的基因表达。在年轻小鼠LUAD肿瘤中显著高表达的基因的同源基因与人类LUAD患者年龄呈负相关。相比之下,在老年小鼠LUAD肿瘤中诱导的同源基因与人类LUAD患者年龄呈正相关(图5c和补充表16)。与年轻或中年患者(<55岁)相比,切除的老年患者(>80岁以上)的人类LUAD组织样本中NUPR1表达更高(图5d和补充表15)。总之,这些发现表明AT2细胞和肺部肿瘤中的与衰老相关的改变在小鼠和人类中显著保守。
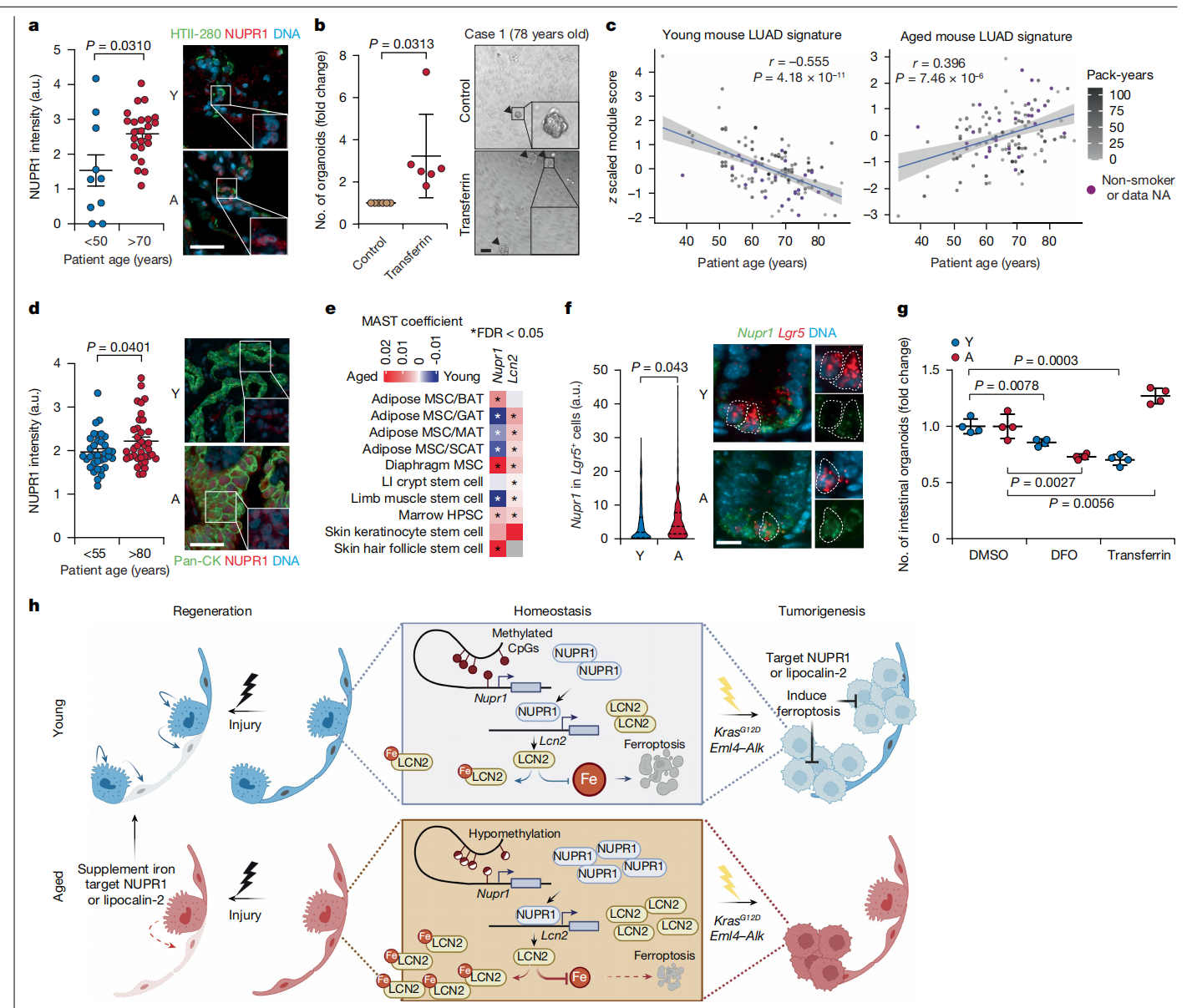
图5:AT2细胞和LUAD中的与衰老相关的变化在人类和其他肺外成人干细胞中保守。
a,老年人(A;n = 24)和年轻或中年人(Y;n = 10)健康肺组织中的HTII-280+ AT2细胞中的NUPR1。为了清晰,插图中省略了HTII-280(绿色)。b,对转铁蛋白反应形成的肺泡类器官的量化(左)和图像(右)。结果以对照归一化(n = 6)。箭头指示人肺泡类器官。c,年轻和老年小鼠LUAD基因表达签名与KRAS突变驱动的LUAD患者年龄的相关性。回归线和95%置信区间已显示。NA,不可用。d,老年人(n = 35)和年轻及中年患者(n = 36)人类LUAD组织(pan-CK+)中NUPR1的量化(左)和图像(右)。e,其他小鼠成人干细胞中的年龄相关变化40,41。数值FDR值列在补充表18中。BAT,棕色脂肪组织;GAT,生殖腺脂肪组织;HPSC,造血干细胞;LI,大肠;MAT,肠系膜脂肪组织;MSC,间充质干细胞;SCAT,皮下脂肪组织。f,定量(左)和图像(右)Lgr5+小鼠肠道干细胞中Nupr1 mRNA的原位杂交(n = 291和286 Lgr5+细胞分别来自3只老年和3只年轻小鼠)。虚线指示Lgr5+细胞。g,DFO或转铁蛋白处理后的肠道类器官形成(n = 4个技术重复,来自1个代表性实验,重复两次)。h,图形摘要。衰老通过表观遗传诱导NUPR1–脂钙蛋白-2轴抑制肺部肿瘤发生和再生,导致老年肺干细胞和肺肿瘤细胞功能性铁不足和对铁死亡的抵抗。比例尺为50 µm(a-d)或20 µm(f)。数据显示均值±标准差(a,b,d,g)。显示中位数和第25及第75百分位数。
讨论
我们展示了与年龄相关的肺AT2细胞干细胞性下降与肿瘤启动潜力的减少密切相关。这一结果直接证明了一个模型,即与年龄相关的干细胞性损失在肺部(可能还有其他组织)具有肿瘤抑制作用,阻止了那些获得致癌突变但干细胞性低于启动肿瘤所需阈值的细胞发生转化。因此,我们的结果可能解释了在老年个体(80-85岁)中观察到的癌症发病率下降,包括肺癌。结合我们的结果和他人的工作表明,50-70岁个体癌症发病率的增加是由于相对年轻个体中的前恶性病变引起的,这些个体的干细胞具有较高的肿瘤生成潜力。这些初始病变最终可能由于突变积累以及在衰老过程中形成的允许性(免疫)微环境和全身代谢变化而进展为临床上可检测的疾病。事实上,在年轻人和中年人的尸检中常规检测到前恶性病变,而在年轻患者中检测到的初始致癌事件已被链接到晚年的肺癌发展。结合我们的结果,这一发现意味着旨在预防和早期干预癌症的努力可能最好针对年轻个体的关键窗口期,此时干细胞最容易受到转化。为了完善这一模型,未来的研究应解决年轻时获得的初始致癌事件是否能保护起源细胞免受与年龄相关的衰退。
我们的工作将铁稳态的表观遗传重编程视为AT2细胞干细胞性和肿瘤生成潜力损失的机制。NUPR1–脂钙蛋白-2轴被认为在感知和控制细胞铁水平以及保护细胞免受铁死亡方面发挥作用。这种平衡在肺上皮中可能特别微妙,因为相对于大多数组织,它长期暴露于高水平的氧气中。我们展示了衰老通过过度激活NUPR1–脂钙蛋白-2轴来打破这种平衡,导致AT2细胞功能性铁不足和对铁死亡的抵抗。我们证明老年干细胞对铁死亡的抵抗对于再生医学和器官移植中使用铁死亡抑制剂以及在癌症治疗中使用铁死亡诱导剂具有重要意义。增加包括p53在内的肿瘤抑制因子的活动可以促进与年龄相关的干细胞衰退,值得注意的是,Nupr1是AT2细胞中p53的目标。因此,与年龄相关的应激输入增加(包括肿瘤抑制因子活动)和表观遗传上许可的低甲基化状态的结合,为老年干细胞中Nupr1的过度激活提供了机制解释。
铁-硫簇作为许多必需酶的关键组成部分,其可用性是高氧环境中肺癌生长的限制因素,这表明与年龄相关的铁不足抑制AT2自我更新和肿瘤发生的一个机制。铁也是一系列酶反应的必要辅因子,包括组蛋白和DNA的去甲基化过程——这些过程对于细胞分化至关重要,包括癌症进展中的细胞状态转变。我们的发现表明,铁对于定义肺癌进展的癌细胞状态转变是必需的。此外,我们展示了老年AT2细胞在受伤后严重阻碍向AT1细胞的分化,通过靶向NUPR1可以挽救这一过程。因此,AT2细胞干细胞性的两个方面,自我更新和分化,都直接受到与年龄相关的功能性铁不足的抑制,这是由NUPR1–脂钙蛋白-2轴的过度活动引起的。我们的发现表明,抑制NUPR1–脂钙蛋白-2轴或补充铁可能是再生医学中恢复老年AT2细胞以及其他成人干细胞类型的有希望的策略。相反,与年龄相关的NUPR1–脂钙蛋白-2轴的诱导可能在广泛的成人干细胞中具有肿瘤抑制作用(图5e)。最后,我们注意到,尽管与年龄相关的NUPR1—脂钙蛋白-2轴的诱导在AT2细胞中抑制了干细胞性和肿瘤发生,但NUPR1和脂钙蛋白-2可能在晚期癌症进展中通过促进老年人的铁死亡抵抗赋予生长优势。
我们在AT2细胞和LUAD细胞之间展示了共享的年龄依赖性基因表达变化,这表明功能性重要的与年龄相关的改变可能被LUAD肿瘤从其起源处表观遗传地继承。我们的结果指出,这种与年龄相关的继承是塑造表观遗传背景的重要力量,在这种背景下获得了致癌突变,从而改变了起源细胞的致癌能力和新兴癌症的分子特征,这是机体年龄的函数。这些与年龄相关的差异可能有助于基于患者年龄个性化癌症预防和治疗策略。
转载本文请联系原作者获取授权,同时请注明本文来自孙学军科学网博客。
链接地址:https://wap.sciencenet.cn/blog-41174-1463542.html?mobile=1
收藏