
 精选
精选
文章引言
2022 年 5 月 19 日,北京大学物理学院、人工微结构和介观物理重点实验室、国家生物医学成像科学中心、北大清华联合生命科学中心、北大定量生物学中心毛有东教授团队在 Nature 杂志发表了去泛素化酶 USP14 调控人源蛋白酶体功能的动力学重编程的重大研究进展,解决了困惑了三十年之久的泛素蛋白酶体的内在调控机制难题,为靶向去泛素化酶的癌症疗法建立了关键分子基础。由于去泛素化酶 USP14 调控人源蛋白酶体的过程具有高度动态和非平衡等动力学特征,传统单颗粒冷冻电子显微镜技术在这项研究中无能为力。为了突破这一技术瓶颈,毛有东教授团队将时间分辨冷冻电镜技术和自主开发的 AlphaCryo4D 人工智能动力学重建技术相结合,实现了对 USP14 调控人源蛋白酶体的完整功能周期的非平衡动力学过程的全原子重建,揭示了 USP14 调控蛋白酶体核酸马达的重编程机制。近期,毛有东教授团队在 International Journal of Molecular Sciences (IJMS) 期刊发表了一篇研究论文,首次详细介绍了其自主开发的 AlphaCryo4D 深度流形学习算法技术框架,及其深入系统的测试和一系列关键应用分析,通过挖掘单颗粒冷冻电镜数据,展示了具有高度构象异质性生物大分子的近原子分辨率的构象空间,实现原子水平生物大分子热力学与动力学特征的机器学习。该研究对于分析生物大分子动力学及其高分辨重建具有重要意义。

AlphaCryo4D 方法框架中的三维深度残差网络模块与赝能量曲面流形学习过程。
研究过程与结果
细胞中重要的生物大分子通常具有复杂的构象异质性特征,如何在对其构象空间进行可视化的同时,仍能突破其分辨率的限制,是冷冻电镜技术发展的核心问题之一。AlphaCryo4D 方法将三维深度残差网络与赝能量曲面的流形嵌入相结合,通过基于能量的颗粒投票策略,从而提高了构象分类的准确度与结构重建的分辨率。AlphaCryo4D 框架主要包括四个计算步骤,第一步通过 M 折颗粒洗牌与极大似然三维分类从单颗粒数据集中采样生成数百至数千个三维体数据,第二步利用深度残差自编码器以非监督的方式进行三维体数据的特征学习,第三步获得三维体数据的低维流形以及颗粒数信息并运用玻尔兹曼分布公式计算赝能量曲面,第四步基于颗粒图像在赝能量曲面上出现的 M个位置 (M 票) 进行投票分类。
在基于模拟数据的算法测试中,AlphaCryo4D 成功构建了分子量为 130 kDa 的 NLRP3 炎性小体 20 个准连续构象变化的赝能量曲面,同时利用 cryoSPARC 中基于概率 PCA 的 3DVA 方法、基于深度变分自编码网络的 cryoDRGN 程序与基于极大似然的 RELION 软件处理相同的模拟数据集进行比较,发现 AlphaCryo4D 的三维分类的精度约为同类算法的三倍左右,对连续构象变化重建分辨率首次达到 2 至 3 埃范围以内。作者进一步将 AlphaCryo4D 应用于蛋白酶体、核糖体与剪接体的冷冻电镜实验数据分析中,验证了其在探索蛋白分子“隐藏”构象空间的潜在普适性,以及突破动态变化结构域局部分辨率限制上的关键应用。
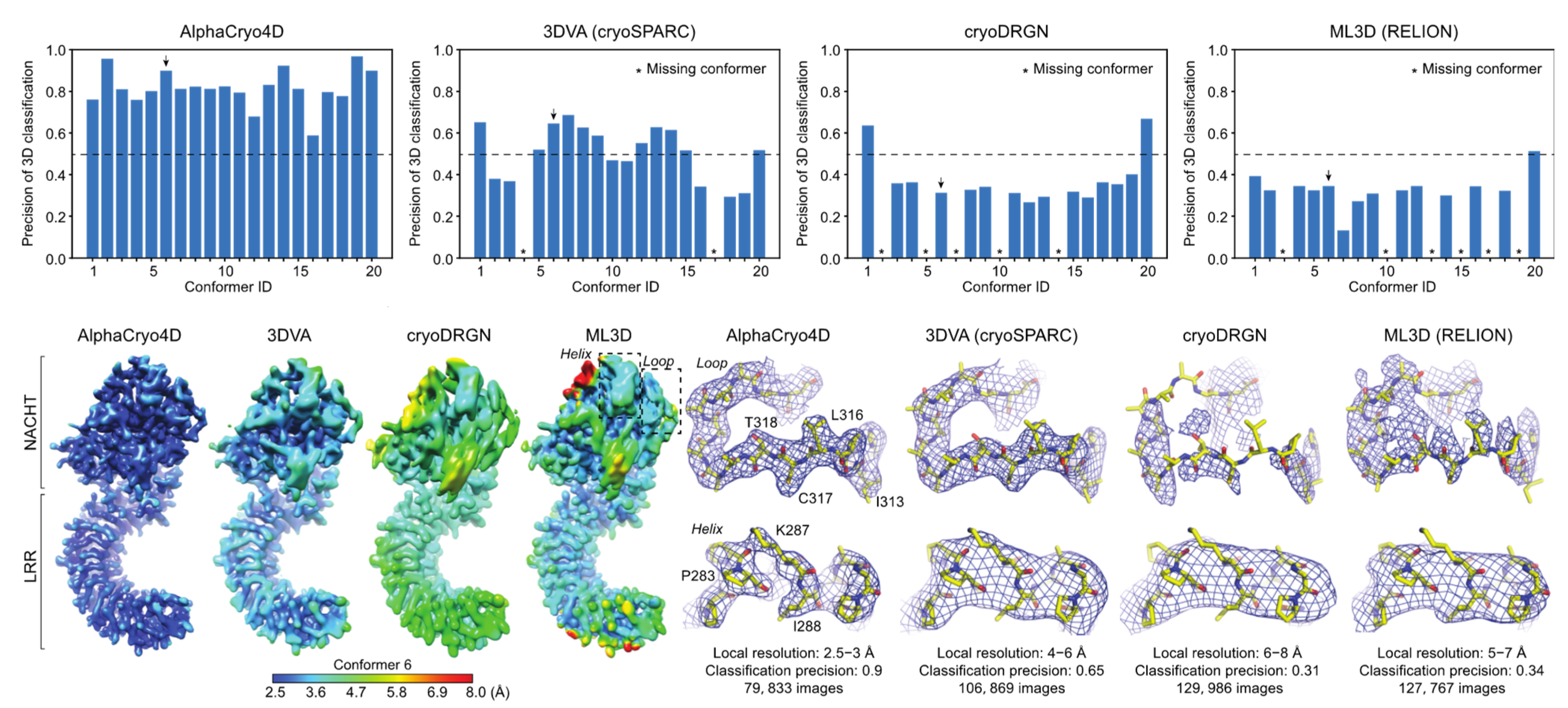
基于模拟数据的其他同类方法 (3DVA、cryoDRGN 和 RELION) 比较验证 AlphaCryo4D 方法在三维分类精度以及重建分辨率上的显著优势。
结论与展望
本研究利用深度流形学习算法,提出了 AlphaCryo4D 方法的整体框架,并通过几个大型的模拟数据与实验数据集初步验证了其在生物大分子构象空间可视化与三维重建分辨率提升方面的适用性和潜在同类应用的普适性。作者在最近的研究工作中,将 AlphaCryo4D 与时间分辨冷冻电镜整合,示范了该技术可用于研究生物分子及复合物处于非平衡状态下发生准连续构象变化的近原子分辨率三维结构,有望在重要药物靶点蛋白应用上揭示“隐藏”或动态配体结合位点的结构机制,从而使以高度动态生物分子为靶点的药物设计成为可能。AlphaCryo4D 方法的研究为冷冻电镜数据中生物分子及复合物近原子水平动态信息的有效提取提供了重要的启示,作者期望 AlphaCryo4D 能够成为冷冻电镜计算方法领域中的一块重要的台阶基石,以激励后续更多的方法学研究,为借助人工智能算法开发下一代冷冻电镜数据处理流程,克服目前面临的难题打下基础,推动原子水平解析生物大分子功能动力学的持续发展。
阅读英文原文:https://www.mdpi.com/1422-0067/23/16/8872
Wu, Z.; Chen, E.; Zhang, S.; Ma, Y.; Mao, Y. Visualizing Conformational Space of Functional Biomolecular Complexes by Deep Manifold Learning. Int. J. Mol. Sci. 2022, 23, 8872.
IJMS 期刊介绍
主编:Maurizio Battino, Marche Polytechnic University, Italy
期刊发表生物化学与分子生物学、生物材料、生物物理、生物医学、化学和纳米科学等分子相关领域研究,已被 Scopus、SCIE (Web of Science)、PubMed 等数据库收录。
2022 Impact Factor:5.6
2022 CiteScore:7.8
Time to First Decision:16.3 Days
Acceptance to Publication:2.6 Days


转载本文请联系原作者获取授权,同时请注明本文来自MDPI开放科学科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3516770-1434868.html?mobile=1
收藏