
代谢学人
Nature:减肥逆袭,从细胞开始变年轻
撰文 | 陈芳芳 胡敏 张俊 武霞 郭明伟
编辑 | 孟美瑶
校对 | 张俊

背景介绍
全球有超过10亿人患有肥胖。脂肪组织(adipose tissue,AT)增大是肥胖的显著特征,也是导致2型糖尿病、心血管疾病、某些癌症和早期死亡的主要危险因素之一。通过体重减轻( weight loss,WL )减少AT质量可显著改善肥胖引起的并发症,并可降低死亡率。要想改善治疗方案和健康状况,就必须对这些截然不同的临床现象的生物学基础有一个协同和详细的了解。
当能量需求发生变化时,AT具有调整自身结构和功能以维持新陈代谢平衡的独特能力。在肥胖症患者中,过度扩张限制了这种灵活性,并诱发病理性重塑改变,特别是脂肪细胞肥大、免疫细胞浸润、促炎细胞因子释放、血管生成受损和纤维化,从而导致多器官炎症、胰岛素抵抗、代谢功能障碍和疾病。然而,尽管进行了广泛的研究,但人们对肥胖AT功能障碍的分子诱因、细胞表型和信号通路仅有部分了解。
治疗性WL可减少主动脉瓣块、全身炎症和胰岛素抵抗,并随后改善肥胖相关的并发症。尽管这有力地表明WL能改善AT功能障碍及其有害的生理效应,但令人惊讶的是,人们对其潜在机制知之甚少。事实上,AT对WL的某些反应可能是不良的,容易导致体重反弹。
想要研究出针对肥胖对健康造成的有害后果的疗法,就必须明确造成病理性和治疗性AT重塑的细胞类型、调节机制和信号通路。

敲黑板啦!
1. 绘制AT重塑动态图;
2. 肥胖AT会导致巨噬细胞持续活化;
3. WL使淋巴细胞浸润减少,增强脂肪细胞代谢灵活性;
4. WL使多细胞应激的逆转,可以抑制衰老。
研究结果
1 .绘制AT重塑动态图
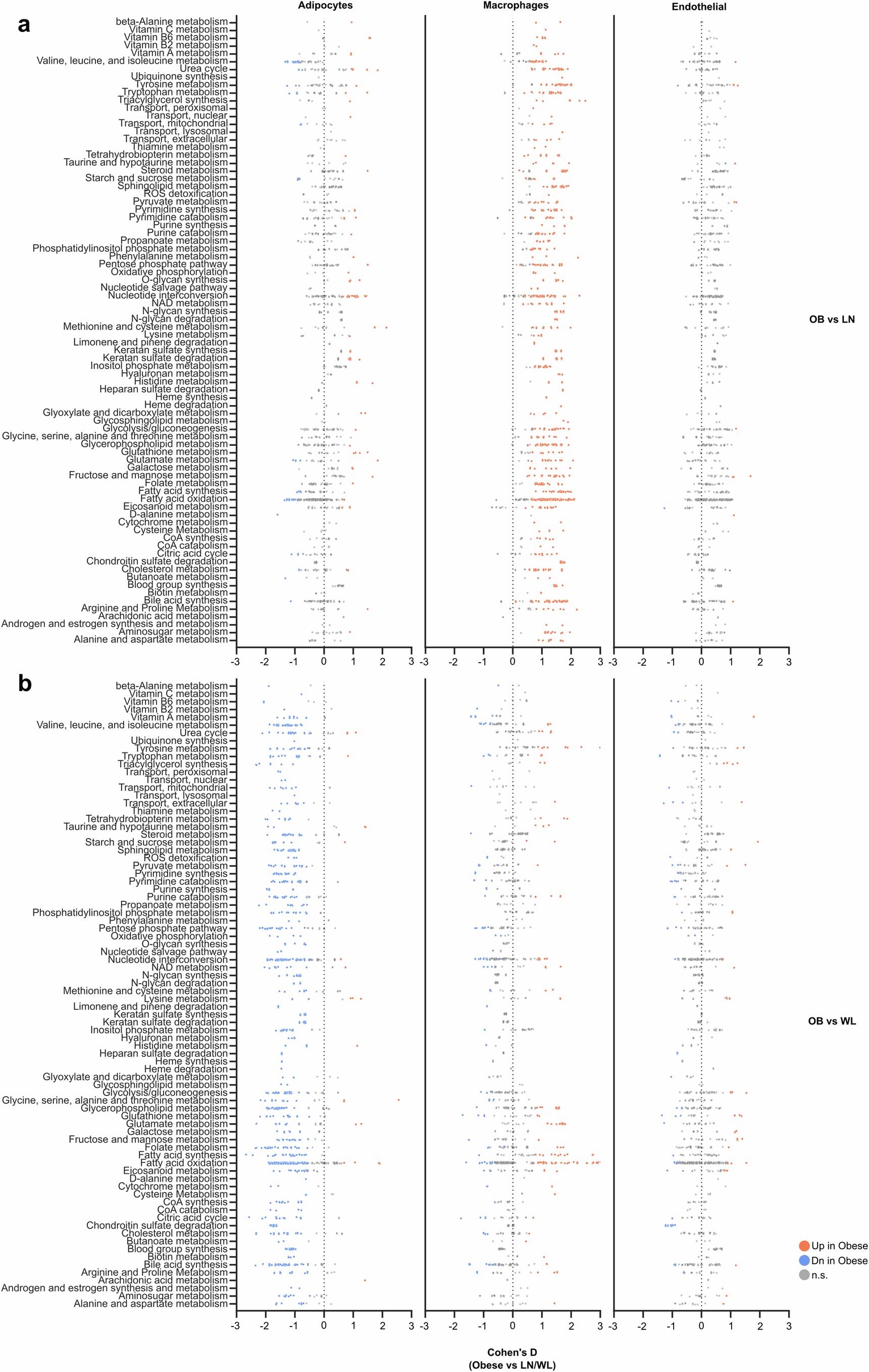
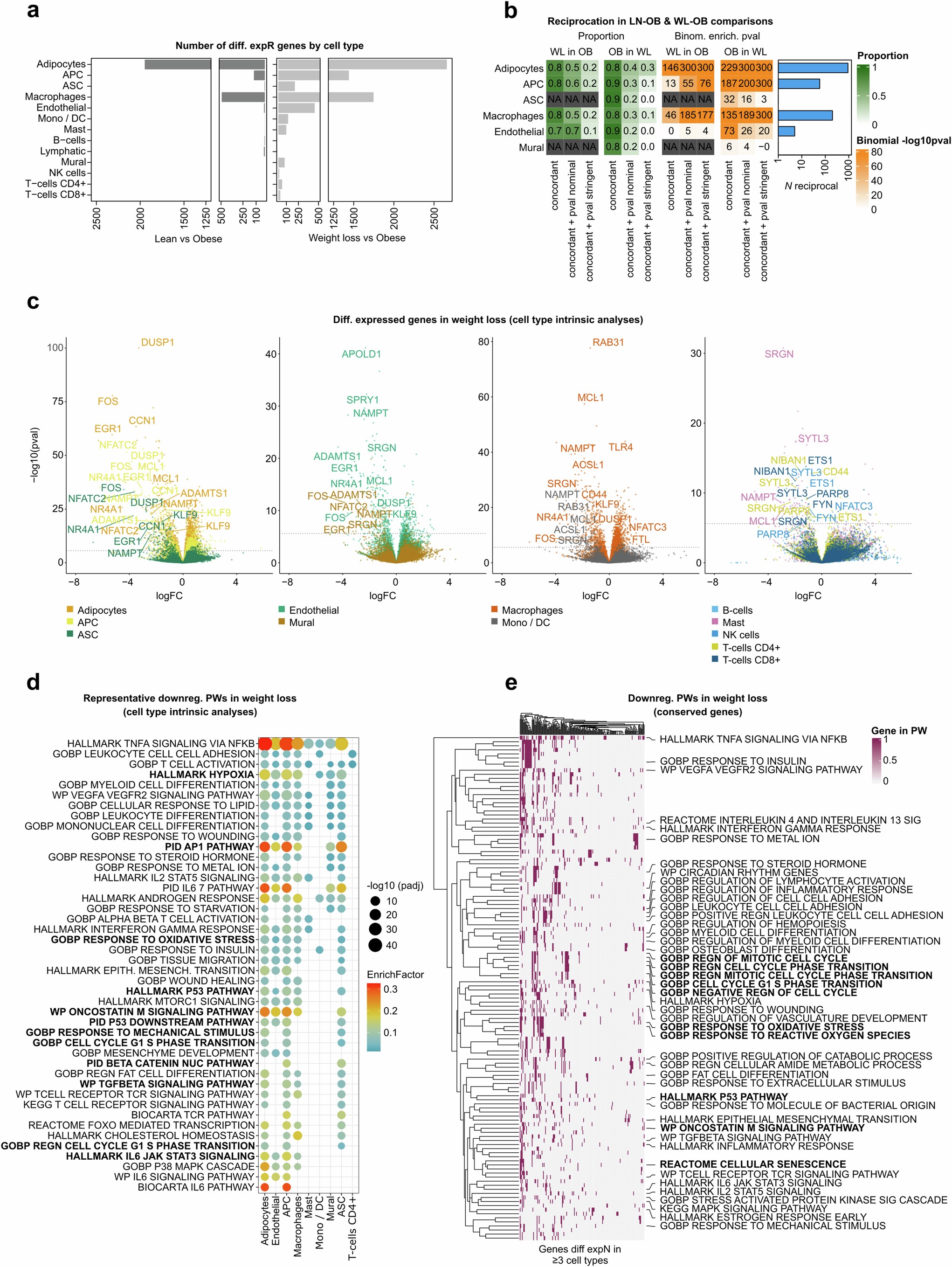
为了更好地了解肥胖AT功能障碍及其在WL术后的逆转情况,研究人员对极度肥胖的男性和女性WL手术前后以及健康瘦的对照组(图1a)的约10万个细胞进行了单核RNA测序。WL能明显改善代谢参数,尽管还达不到瘦人的水平(图1b和扩展数据表1)。研究人员将重点放在腹部皮下脂肪组织上,因为与其他皮下脂肪组织相比,腹部皮下脂肪组织对中心性肥胖有贡献作用,而且对代谢有不利影响(小编注:这里作者的意思是与其他皮下脂肪组织相比,腹部皮下脂肪组织对中心性肥胖有贡献作用。这意味着腹部皮下脂肪的增加也构成了中心性肥胖外观(腰围增大)的一部分,但它不是主要或唯一的驱动因素。腹腔内的内脏脂肪才是核心。文献Michael M. Swarbrick,2014中研究了臀部皮下脂肪组织(gluteal SAT,GSAT)和腹部皮下脂肪组织(abdominal SAT ,ASAT),发现GSAT和ASAT质量与空腹胰岛素血症、胰岛素抵抗和血脂异常(臀部是有益的;腹部是有害的)表现出相反的关系,并且ASAT释放出超出GAST 4倍多的炎症因子,提示相对于其他皮下脂肪组织,腹部皮下脂肪组织对中心性肥胖具有促进作用)。这组样本构成了组间探索性分析的基础。他们将研究结果与已发表的最大人体皮下脂肪图谱中的另外50000个细胞(核)进行了整合,以改进细胞注释(n=9个肥胖样本,n=4个瘦样本;扩展数据图1a-c)。研究人员在等量组群中进行了空间转录组学分析,使他们能够在健康和功能失调AT的组织层次中,对细胞表型进行空间定位和功能定位。(约25,000个细胞,每组n=4。图1a,扩展数据图1b,c和扩展数据表1)。
研究人员利用分析捕捉到了人体体重增加和WL过程中皮下AT微环境的细胞、结构和功能动态的丰富表征。全组织聚类和组成成分分析表明(图1c),肥胖AT中存在广泛的免疫细胞(主要是巨噬细胞,也包括淋巴细胞)浸润(图1d,e和扩展数据图1d,e)。肥胖AT还表现出成熟脂肪细胞的不足,表明细胞死亡增加和/或成熟脂肪细胞补充不足。WL减轻了这些典型的有害影响。
![]()
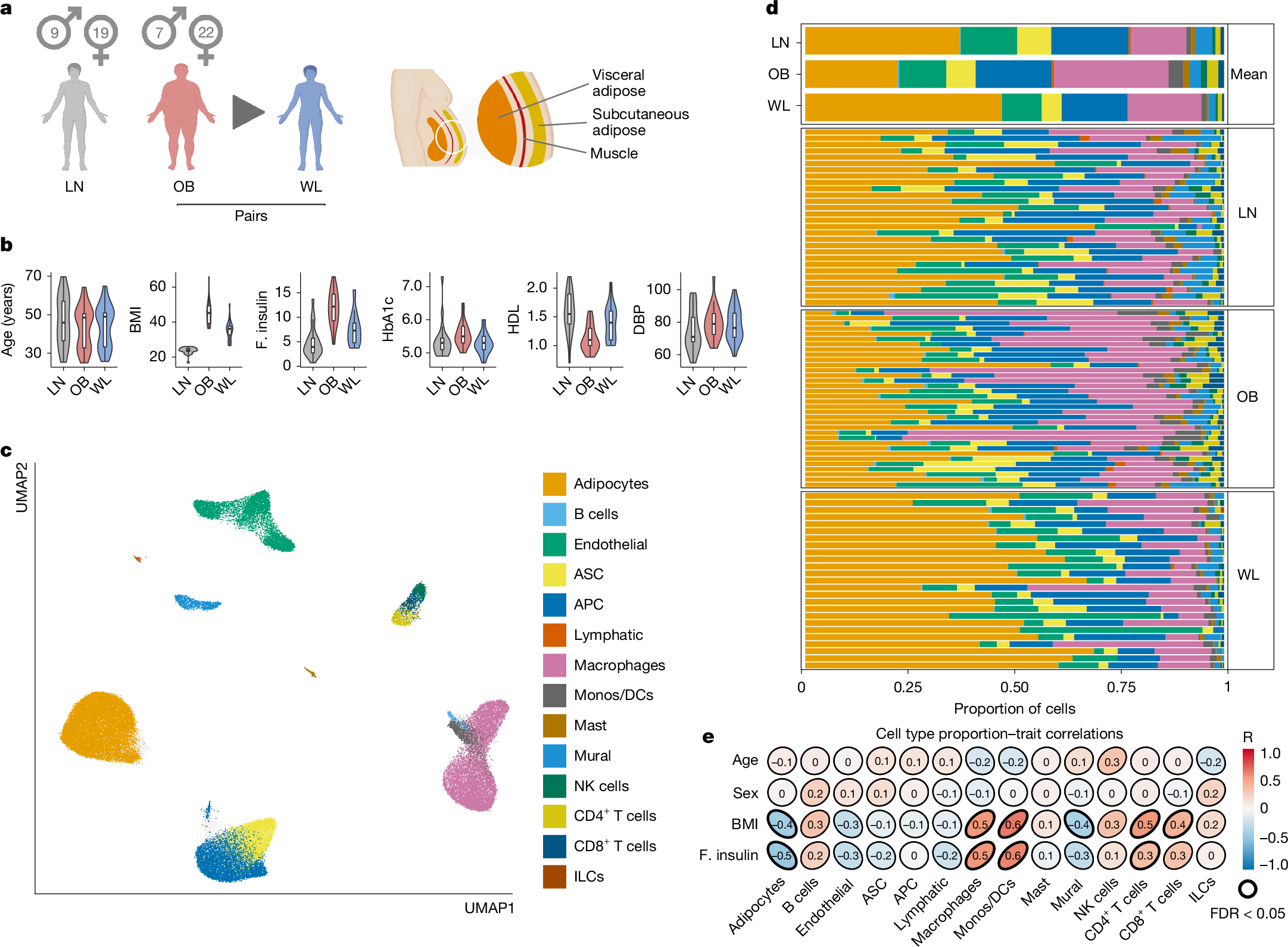
图1 | 人类AT在瘦、肥胖和WL方面的单细胞图谱
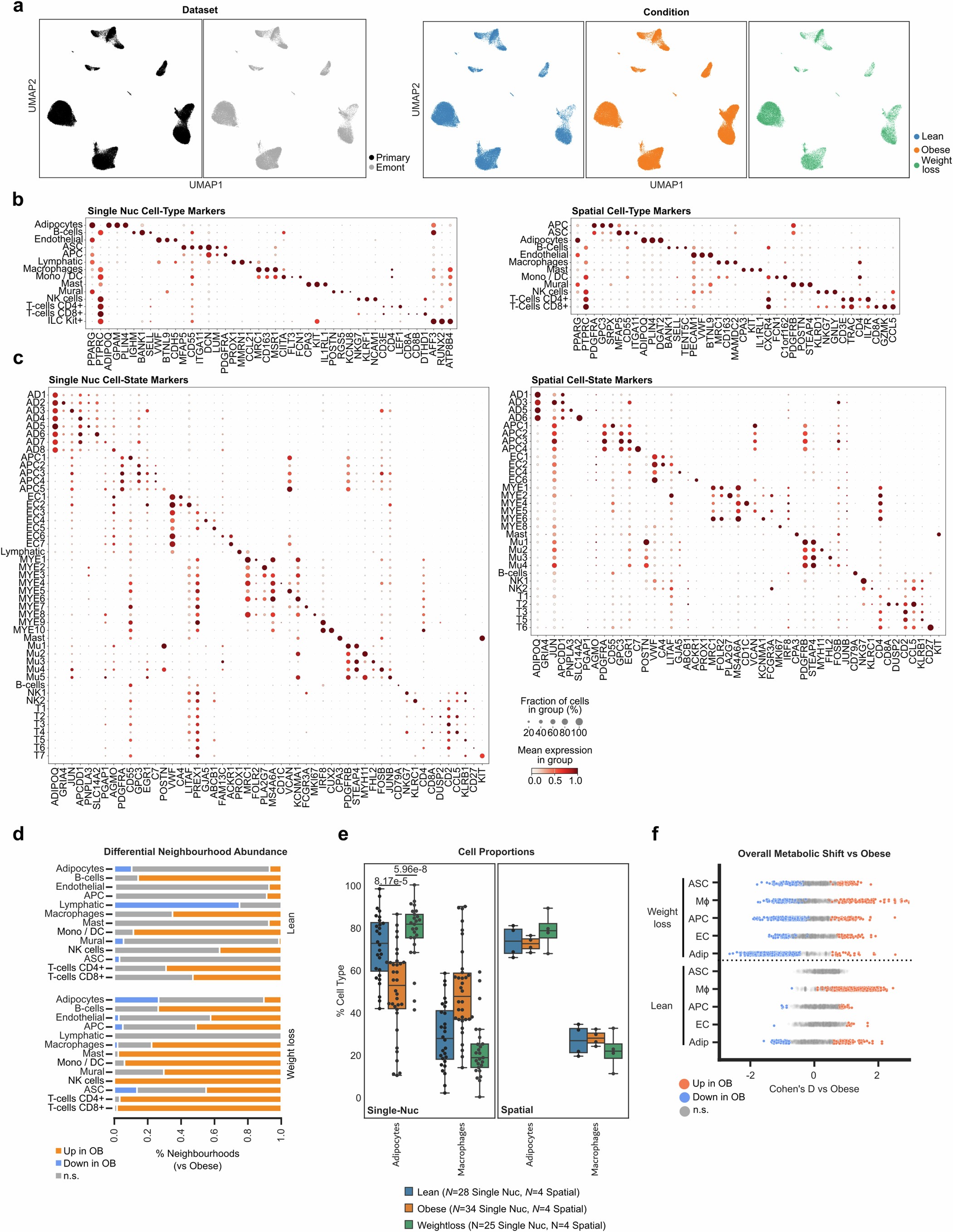
扩展数据图1 | 瘦脂肪组织、肥胖脂肪组织和WL脂肪组织中单核和空间分辨的细胞类型和状态的变化
2 .巨噬细胞持续活化
免疫细胞浸润是肥胖AT的一个特征,但WL对炎症重塑的影响尚不清楚。有研究表明,WL具有抗炎和促炎相反的作用。
扩展阅读:减肥后AT中的炎症重塑
减肥后AT中的炎症重塑呈现复杂的双重性:一方面,减重能显著缓解慢性低度炎症(抗炎作用);另一方面,残留的免疫细胞代谢重编程和微环境变化可能诱发短暂促炎反应,共同影响体重反弹风险。本文中减重显著降低AT中巨噬细胞总数(如研究显示从31%降至18%),尤其减少促炎亚群 LAM ST2(高表达TLR2/TREM1,驱动胰岛素抵抗)。巨噬细胞向修复型(如LAM ST1)偏移,促进脂质清除和组织修复。并且炎症通路被系统性抑制,下调多种关键炎症因子,如炎性体通路NLRP3下调(减少IL-1β释放);趋化因子:CCL3/CCL4下调(抑制单核细胞招募);TLR信号下调(削弱病原识别受体介导的炎症放大),减重逆转脂肪细胞和血管细胞的衰老,减少 SASP因子(如IL-6、TNF-α)分泌。但减重也存在潜在促炎作用,残留炎症记忆与急性应激。如免疫细胞代谢重编程未重置,残留巨噬细胞维持全局代谢激活状态,糖酵解(HK2↑)和磷酸戊糖途径持续活跃,支持细胞存活和炎症潜能。氧化应激基因(如NOX4)高表达,促进活性氧(ROS)积累。肥胖期染色质重塑(如H3K4me3修饰)使炎症基因保持“开放”状态,即使脂质微环境改善,细胞仍处于预激活状态。减重初期脂解加速,大量游离脂肪酸(FFA)涌入微环境,激活巨噬细胞 TLR4通路,诱导TNF-α、IL-6表达。促进 ROS生成 ,激活NLRP3炎性体。临床表现为短暂炎症标志物升高(如CRP),尤其在快速减重(如减肥手术)后。另外部分研究报道减重后 CD8+ T细胞比例升高,分泌IFN-γ放大炎症。脂肪细胞缩小释放 DAMPs(损伤相关分子模式),通过TLR2/4激活残留巨噬细胞。
参考文献:
[1] van Baak, M. A, et al. Nat. Rev. Endocrinol. 2019 Jan 17.
[2] Hinte LC, et al. Nature. 2024 Nov 18.
研究人员将髓系细胞分为AT巨噬细胞、单核细胞和树突状细胞(MYE1-10)等异质性亚类(n=34280)(图2a,扩展数据图2a和补充表2)。AT巨噬细胞的增加(平均从14%到31%)主要包括表达溶酶体、脂质代谢和代谢活化标志物(CD9、TREM2、LPL、LIPA)的脂质相关巨噬细胞(LAMs;成熟的MYE2和不成熟的MYE3)(图2a-b和扩展数据图2a-c)。表达VCAN的经典单核细胞(MYE5)也增加,这表明它们是从血液中组成性迁移而来的(小编注:VCAN(Versican,多功能蛋白聚糖)是一种大分子的细胞外基质蛋白多糖。它在炎症、细胞粘附、迁移和信号传导中发挥作用。单核细胞表达VCAN通常与它们的激活状态、迁移能力或特定功能亚群有关。VCAN+MYE5细胞处于迁移-分化过渡态,具备迁移能力但尚未获得完全促炎功能。MYE5细胞不表达典型炎症因子(如 TNF、IL1B)(图2b),区别于促炎巨噬细胞(如LAM ST2)。MYE5是单核细胞向巨噬细胞分化的中间状态,其VCAN表达反映迁移能力,而非终末炎症功能(如补充表4所示:MYE5高表达 VCAN、SELL(L-选择素),低表达 CD68、MRC1)。VCAN是一种细胞外基质蛋白,在单核细胞穿越血管内皮时高表达,介导细胞与内皮黏附及跨内皮迁移(Transendothelial Migration)。本研究显示,肥胖AT中 VCAN+经典单核细胞(MYE5)比例显著升高(图2a),空间转录组数据(图2d)进一步显示,VCAN+细胞富集于血管周围,支持其迁移中或刚迁入的状态。)。研究人员通过数据可视化和标记基因模式,描述了从单核细胞到未成熟再到成熟LAMs的连续分化的过程(图2a和扩展数据图2a)。研究人员通过比例分析显示,表达稳态标志物(LYVE1、FOLR2、MRC1)的组织驻留巨噬细胞(TRMs,即MYE1亚群)的比例较低(扩展数据图2c)。邻域图证实了TRM的相对减少(而非绝对)(小编注:邻域图(Neighborhood Graph) 是一种基于细胞空间位置关系的计算模型,用于量化细胞群体的分布密度(绝对数量)和组成比例(相对占比)。这里蜂群图展示了邻域丰度变化(Spatial FDR < 0.1)。横轴是邻域丰度变化(Log2FC),>0表示该细胞比例增加(如OB vs Lean一列);<0表示该细胞比例减少,这里OB vs 减重一列,MYE1(TRM)中蓝点相对于OB vs Lean一列增多,表明TRM的相对减少(而非绝对))(扩展数据图2b)。增殖性巨噬细胞表达MCP-1 (CCL2)、TRM 和 LAM 的标记物,这支持了在人类肥胖中存在 MCP-1 依赖性的两种巨噬细胞群体(TRM 和 LAM)的低水平扩增(扩展数据图2a)。
与脂肪无关,LAM丰度随代谢功能障碍增加而增加(扩展数据图2b)(小编注:这里FI adj BMI 是指空腹胰岛素(FI)校正 BMI(与肥胖无关),专注代谢功能障碍(空腹胰岛素升高)与细胞比例的关系。红点表示空腹胰岛素升高,该细胞比例增加。)。这使研究人员假设LAMs可能具有多效应适应性和不良适应性特征。LAM亚聚类发现了两个主要亚群,它们分别代表溶酶体或代谢(LAM ST1,适应性)和炎症(LAM ST2,不良适应性;MHCⅡ、NLRP3)的特征(图2b)。炎症性LAMs表达较高的TLR2和TREM1,它们是在病原体识别反应中启动和放大炎症反应的协同受体(图2b,扩展数据图2d和补充表5)。与有害作用相一致的是,肥胖症患者的炎性LAM数量增加与代谢功能障碍有关(图2c)。空间和蛋白质分析表明,LAMs的方向和功能与环境有关,适应性LAMs聚集在冠状结构(crown-like structures,CLS;转录缺乏的脂肪细胞周围),而炎症性LAMs在孤立或配对的情况下更丰富(图2d和扩展数据图2e-g)(小编注:这里的配对是指炎症性LAM彼此配对(LAM-LAM pair)分布,而不是与其他细胞类型配对。空间数据提示它们不集中在CLS,而是散在单个或两两相邻的分布模式。)。
为了无偏向地了解巨噬细胞的代谢重编程,研究人员使用基因表达对代谢通量进行了系统的建模(小编注:研究者使用转录组数据(基因表达水平)结合已知的代谢网络模型(例如Recon或HumanGEM),推算各代谢反应的相对活跃程度,即代谢通量。这种方法通过代谢酶编码基因的表达量来估计代谢反应的流速和方向,从而推断细胞整体代谢状态变化。)。这揭示了肥胖巨噬细胞所特有的一种全局性激活状态,包括已知的和以前未被发现的代谢变化(图2e,扩展数据图1f和3a,b及附表6)。具体来说,研究人员在肥胖小鼠中发现了与细胞外通量分析(小编注:细胞外通量分析(extracellular flux analysis)是一种代谢功能检测方法(如Seahorse XF分析),测量的是细胞在培养环境中消耗氧气(OCR)和产生酸(ECAR)的速率,分别反映线粒体呼吸和糖酵解活性。这虽然是外部测量,但反映的正是细胞内的代谢功能状态。)一致的高糖酵解(促炎)、高呼吸作用(抗炎)模式并存的代谢模式的转变(小编注:在肥胖巨噬细胞中,这两种模式是同时增强的,而不是从一种转变为另一种。即既有促炎的高糖酵解,又有抗炎的高呼吸活性,这是一种“双高”状态。);以及磷酸戊糖途径和TCA循环的活性增强(小编注:戊糖磷酸途径(PPP)和TCA循环均表现为活性增强,与能量代谢和生物合成需求上升一致。);胆固醇、脂质和脂肪酸合成及氧化途径的广泛激活;细胞转运的强制性上调(图2e,f和扩展数据图3a)。以脂肪酸为例,通量模型揭示了脂肪酸去饱和(FADS1和SCD)和线粒体β-氧化的显著激活,这与缓冲和利用具有潜在毒性的微环境脂肪酸作为能量是一致的(小编注:脂肪酸去饱和(FADS1、SCD)将饱和脂肪酸转化为单/多不饱和脂肪酸,不饱和形式更易被储存或氧化,从而降低饱和脂肪酸的脂毒性,并为膜合成和信号分子生成提供底物。)(图2e,f)。全局性激活作用在LAMs中最为明显,但并不局限于LAMs(扩展数据图 2h),这表明肥胖AT 中的各种髓系类群都经历了广泛的新陈代谢启动。实验能量谱分析证实,LAMs比TRMs具有更高的基础活性和糖酵解能力,这证实了他们基于转录组的通量结果(图2g,扩展数据图2i)。
WL导致各亚类髓系细胞数量明显减少(平均从31%到18%)(扩展数据图2b)。比例和密度分析显示,部分髓系细胞比例在肥胖和WL之间没有差异(图2a和扩展数据图2c),研究人员在原位中验证了这一点(扩展数据图2j)。然而,WL确实使LAMs向炎症较轻的亚型转移(图2c)。总之,这表明尽管有大量的WL,肥胖诱导的髓细胞状态仍然存在。转录组通量分析证实,全局性代谢活化并没有随着WL而完全逆转(图2e和扩展数据图3b)。但是WL的确显著降低了脂肪酸合成和氧化(主要为去饱和酶和酰基辅酶A合成酶)(小编注:去饱和酶是脂肪酸合成途径的关键环节之一,把新合成或已有的饱和脂肪酸转化为不饱和脂肪酸;而这些不饱和脂肪酸既可用于储存(甘油三酯合成),也可进入脂肪酸氧化途径供能,因此它在合成和氧化两端都有作用。)的某些环节,从而在时间上将这些途径与微环境脂质供应联系起来(小编注:WL会减少组织中的游离脂肪酸水平,因此细胞对这些脂肪酸的加工需求下降,对应去饱和酶(加工脂肪酸)和酰基辅酶A合成酶(活化脂肪酸进入代谢途径)的表达也下降。这说明这些酶的高表达在肥胖时是为了应对脂质过剩,而在WL后随着脂质供应减少,代谢加工负荷也随之降低)(图2e-f)。相反,糖酵解、呼吸能力和磷酸戊糖途径通量增加了(图2e-f),这意味着随着脂肪酸水平的降低,需要从其他来源获取能量。差异表达分析显示,炎症小体、促炎细胞因子和趋化性基因广泛减少(图2f,扩展数据图2k和补充表7和8)。网络分析表明,特定的转录因子(TFs)参与了TRM和LAM的正常化表达,并揭示了WL可改善炎症和稳态网络的模式,但不能改善LAM转录重编程(图2h及附表9和10)。总之,这些结果表明,肥胖AT中存在着以单核细胞招募和持续代谢重编程为主的复杂激活反应。
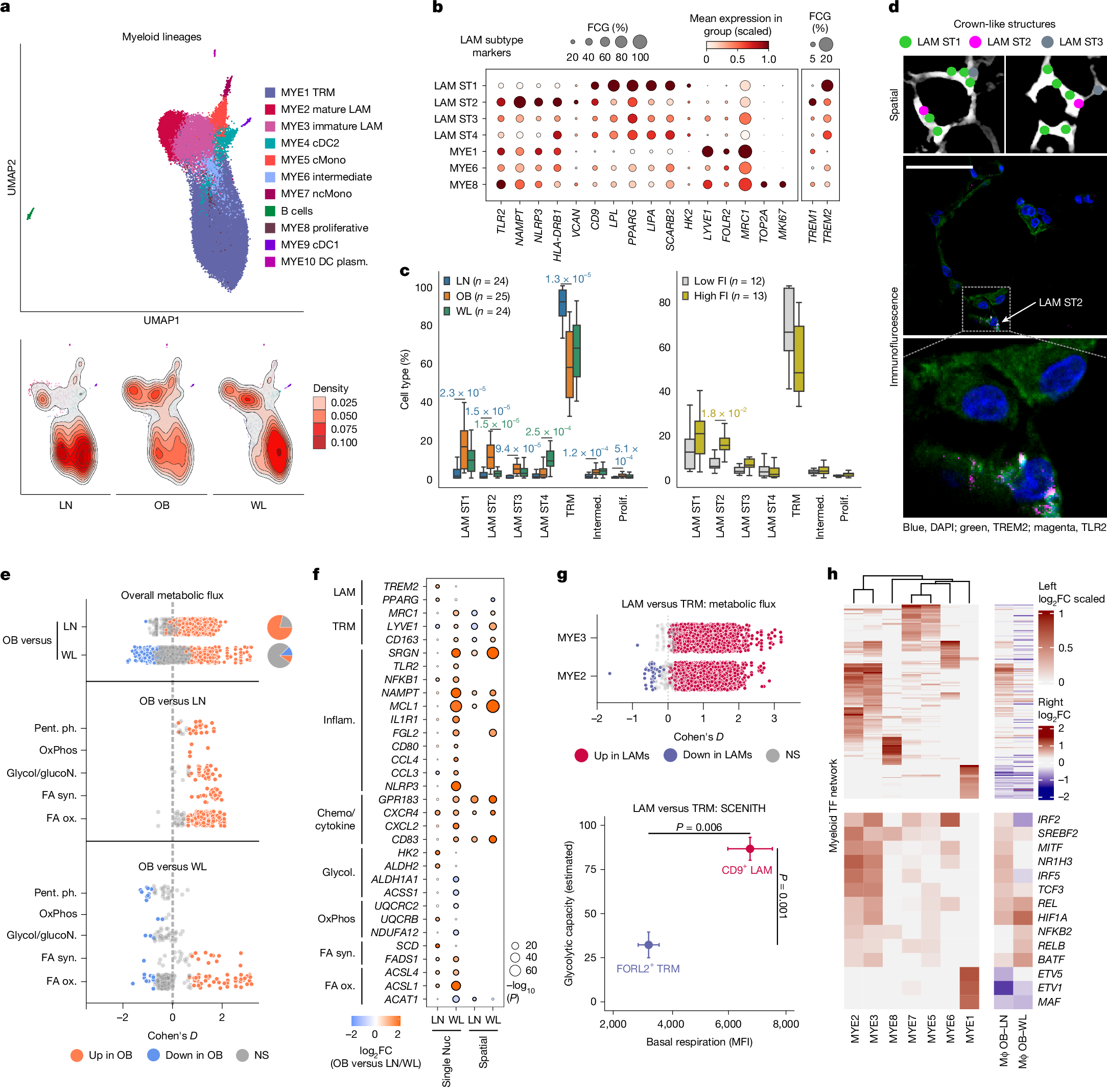
图2 | 肥胖和WL中的免疫细胞浸润、激活和重编程
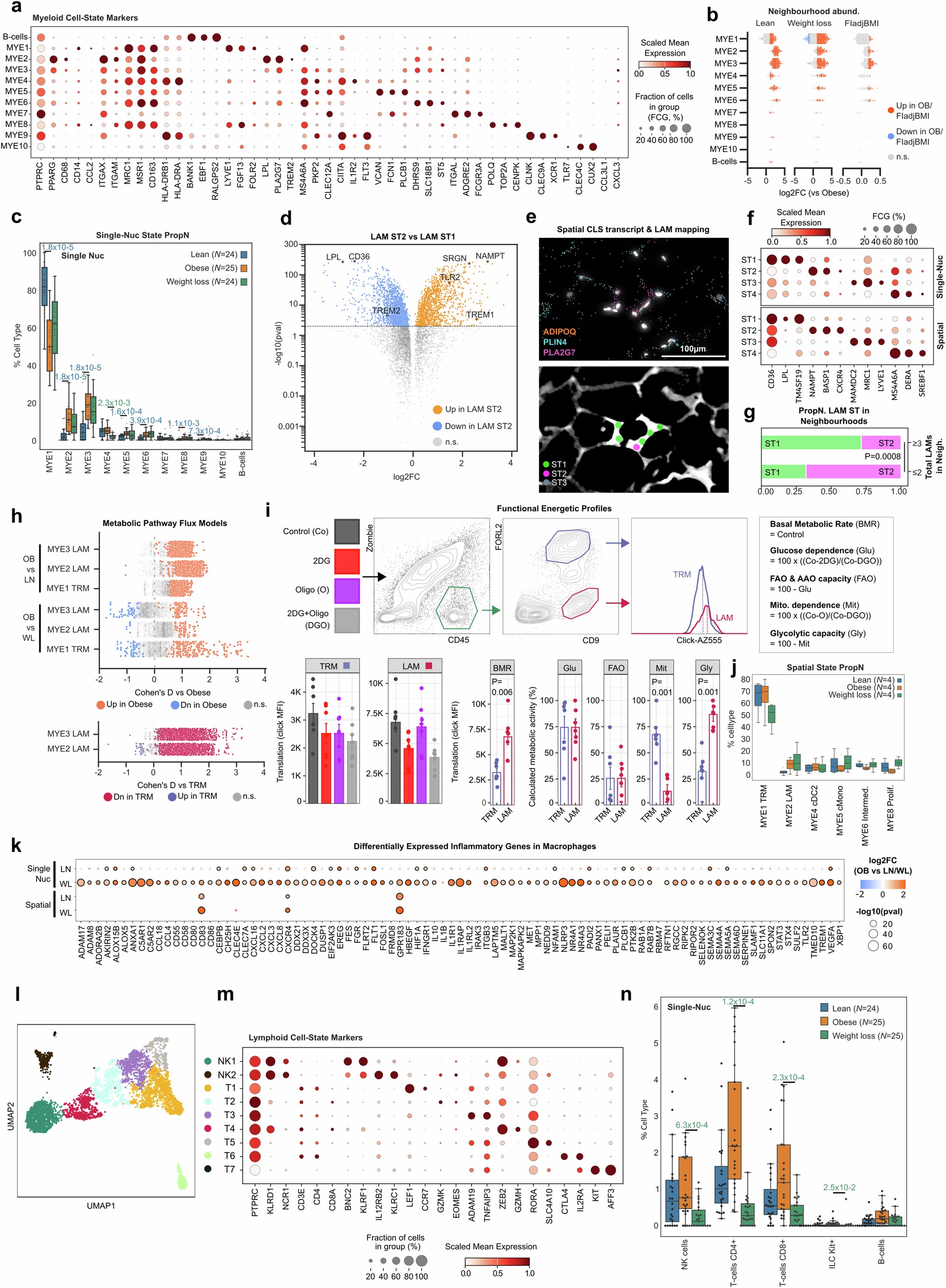
扩展数据图2 | 脂肪组织免疫系统在人体体重增加和WL变化中的作用
扩展数据图3 | 成熟脂肪细胞和巨噬细胞983条途径,1895个反应代谢通路全谱流量变化
3 . 淋巴细胞浸润减少
总体细胞数量较少(6222(4%))意味着研究人员无法评估淋巴亚类水平的变化(扩展数据图2I-n)。然而,肥胖AT具有更高的CD4+和CD8+T细胞、NK细胞和B细胞比例,WL可改善其重塑效果(扩展数据图2n)。WL还下调了淋巴细胞活化和细胞毒性基因(ETS1和SYTL3),进一步支持了炎症的降低(补充表7和8)。
4 .增强脂肪细胞代谢灵活性
成熟的脂肪细胞在肥胖和WL过程中会经历多种表型变化,通过扩张和收缩来适应不断变化的能量需求,然而尚不清楚这些变化如何影响脂肪细胞的分子特征和代谢功能。研究人员通过对成熟脂肪细胞进行进一步的聚类,发现8个细胞亚群(AD1~AD8,n=44583个细胞)(图3a,扩展数据图4a-b和补充表2)。两个亚群分别表现出“应激型”(AD3,JUN/NFKBIZ-hi)和“纤维化型”(AD6、NOX4/LOX-1-hi)特征。这两个亚群随着肥胖而增加,表明这是一种响应组织微环境的选择性变化(图3b)。另一个具有脂质生物合成特征的亚群(AD5,PNPLA3/MOGAT1-hi)在肥胖中意外地减少(图3b)。相反,WL导致应激脂肪细胞显著减少(从55%减少到14%),纤维化细胞数量减少,脂质生物合成细胞相对增加(图3b)。应激和脂质生物合成亚群的变化在mRNA原位杂交中得到了验证(扩展数据图4c)。米色脂肪细胞是罕见的(AD8 GATM-hi,1%),并且在条件之间保持不变。
基于表达的代谢通量分析发现,与瘦脂肪细胞相比,肥胖脂肪细胞的脂肪酸和支链氨基酸(BCAA)分解存在显著缺陷,这与之前的研究结果一致,共同表明代谢灵活性受损(图3c和扩展数据图3a)。相比之下,WL导致脂肪细胞代谢通量显著增加,可能反映了能量负平衡(图3c,扩展数据图1f、3b及附表6)。
由于WL会导致合成代谢增加,于是作者提出假说:热量限制触发的甘油三酯动员可能启动了脂质循环(lipid cycling),即甘油三酯的重复降解与再合成,该过程是对热量限制的一种生理反应。为了验证通量模型,研究人员比较了各组中重要底物通路的酶活性和通路限速酶表达水平(图3d-e和扩展数据图4d-f)。 肥胖脂肪细胞始终表现出较低的代谢活性,再次证实其代谢灵活性受损。WL系统性激活了脂质生物合成和分解这对拮抗通路(图3d)。与此一致的是,研究人员发现脂质循环中甘油三酯分解与再合成的生化反应级联中的典型酶发生了显著变化,包括DGAT2,它编码一种酰基转移酶,可催化甘油三酯的合成并介导脂质循环(图3e)。由于酶的表达是决定催化能力的关键因素,所以WL可能启动了甘油三酯循环,这是一个高生物能的过程,具有重要的脂质多样化和淬灭有毒脂肪酸的代谢益处。WL还逆转了BCAA分解代谢的缺陷,表现为BCAA分解代谢和胰岛素敏感性都得到明显改善(图3c-e和扩展数据图4f)。脂质循环是PNPLA3-hi脂肪细胞(AD5)的特征,而应激(AD3)脂肪细胞的特征是代谢周转率低(图3f)。总之,WL不仅激活经典分解代谢,还增强了细胞自主性的脂质循环,这可能是其广泛改善代谢稳态的基础。
扩展阅读:甘油三酯循环
甘油三酯循环是一种多步骤的无效循环途径,涉及脂肪细胞内甘油三酯(TAG)的持续水解与再酯化:正向反应(脂解):TAG被脂肪酶(如ATGL、HSL、MGL)逐步水解为甘油和游离脂肪酸(FFAs)。反向反应(再酯化):水解产生的FA在消耗ATP的条件下,被重新酯化为TAG(通过DGAT1/2等酶催化)。该过程属于三大UCP1非依赖产热循环之一的甘油三酯-脂肪酸无效循环(TAG-fatty acid cycling)。详情请参阅我们此前的推文:“代谢典藏 | 产热脂肪:超越你想象”。此外,还存在一个看似相似的概念:脂肪酸从头合成-氧化(FAS-FAO)无效循环。该过程起始于各个来源的乙酰CoA,通过三种重要的脂肪酸合成酶(ACLY、ACC和FASN)催化形成FA。新合成的FA一方面可以与甘油发生酯化而形成TG,进一步进入上述的TG循环;另一方面可以进入线粒体被氧化,于是形成了FAS-FAO无效循环,该循环由于无ATP净生成,故不直接用于产热,而是抑制TCA循环过载,进而调节UCP1依赖的产热。详情请见往期推送:“Nature Metabolism:内耗也能发热,褐脂不讲武德”。下图详细展示了上述的TAG循环和FAS-FAO循环。

脂质多样化指脂肪细胞通过循环动态调整脂滴中脂肪酸的组成,以适应代谢需求,主要通过以下机制:1、选择性再酯化:再酯化过程中,细胞优先将特定脂肪酸(如多不饱和脂肪酸PUFA)掺入新生TG,替换原有饱和脂肪酸(SFA)。2、避免SFA积累:SFA易引发脂毒性,循环通过将SFA替换为不饱和脂肪酸(PUFA),减少其细胞毒性,即降低内质网应激和炎症。3、重塑脂滴组成:通过持续水解-再酯化,脂滴的脂肪酸谱更适应能量需求(如寒冷时增加PUFA以促进流动性)。TG循环是能量稳态的核心调节器:在产热上,TG循环形成的ATP可直接水解,用于产热(约占总能量消耗的10%),是UCP1非依赖性产热的重要途径。在调节代谢灵活性上,该循环能平衡脂解(供能)与再酯化(储能),快速响应能量需求变化(如寒冷/饥饿)。此外,该循环还能起到代谢缓冲作用,与肝脏的甘油-葡萄糖循环和肌肉的乳酸循环(Cori循环)联动,维持全身代谢稳态。
参考文献:
[1] Sharma AK, Khandelwal R, Wolfrum C. Cell Metab. 2024 Jun 4;36(6):1184-1203.
[2] Cohen P, Kajimura S. Nat Rev Mol Cell Biol. 2021 Jun;22(6):393-409.
为了明确哪些TFs负责WL诱导的代谢激活,研究人员进行了仅限于代谢通路基因的网络分析(扩展数据图5g和补充表11)。MLXILP和SREBF1(小编注:二者均调控甘油三酯合成基因。1、MLXILP编码蛋白与MAX样蛋白X (MLX)形成异源二聚体,发挥激活转录的作用,并参与响应细胞葡萄糖水平的基因调节。此外,在脂肪代谢方面,MLXILP可以促进脂肪酸合成相关基因的表达。2、SREBF1(固醇调节元件结合蛋白1)是一种基本的螺旋-环-螺旋亮氨酸拉链转录因子。当细胞内胆固醇等脂质水平较低时,SREBF1会被转运到细胞核,在核内与固醇调节元件(SRE)结合,启动这些脂质合成相关基因的转录,从而促进脂肪酸和胆固醇的合成,以满足细胞膜合成和能量储存等需求。)在甘油三酯合成中排名很高,说明它们与调控WL诱导的脂质循环有关。此外,研究人员还发现许多与氧化还原和BCAA分解代谢相关的TFs。53个显著TF中,38个与人类代谢性疾病GWAS位点重叠(扩展图4g),提示这些TF及其调控通路可能在这些疾病病理和治疗响应中具有调控作用,或者这些TF的变化继发于这些疾病(扩展数据图4g)。
差异表达分析发现,生物力学的改变是脂肪细胞应激和代谢功能障碍的潜在驱动因素,而WL减轻了这一现象。具体而言,肥胖增加了细胞骨架张力、机械传导、细胞外基质(ECM)形成和纤维化基因(ACTA2、LOX、LOXL2和VGLL3)的表达,而WL下调上述基因并缓解代谢功能障碍。这些基因在应激/纤维化脂肪细胞(AD3/AD6) 中富集(图3g,扩展数据图4h及附表7和8)。因此,研究人员评估了脂肪细胞肥大和机械应力是否会引发这些不良变化,以及WL期间的脂肪细胞缩小是否可能逆转这些变化。结果显示,脂肪细胞体积在肥胖时增加,而在WL期间体积缩小(图3h和扩展数据图4i)。尽管样本间存在异质性,但脂肪细胞大小与机械敏感基因、应激和纤维化基因的表达呈正相关,而与稳态基因呈负相关(图3g,以图3h中的应激标记物JUN为例)。总之,以上结果提示机械应力是组织退行性循环的关键驱动因素之一。
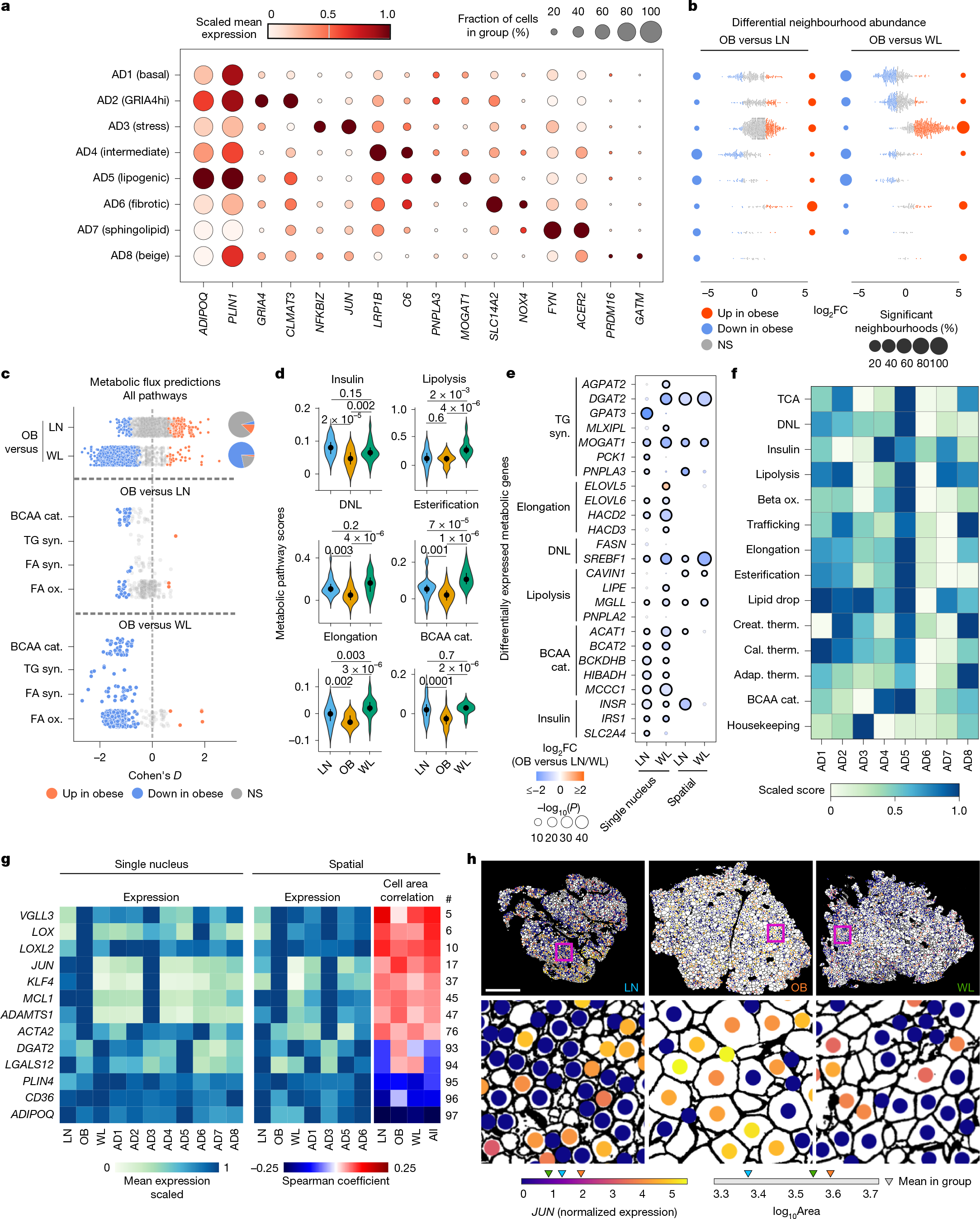
图3 | 肥胖和WL中脂肪细胞和分子谱的动态调节
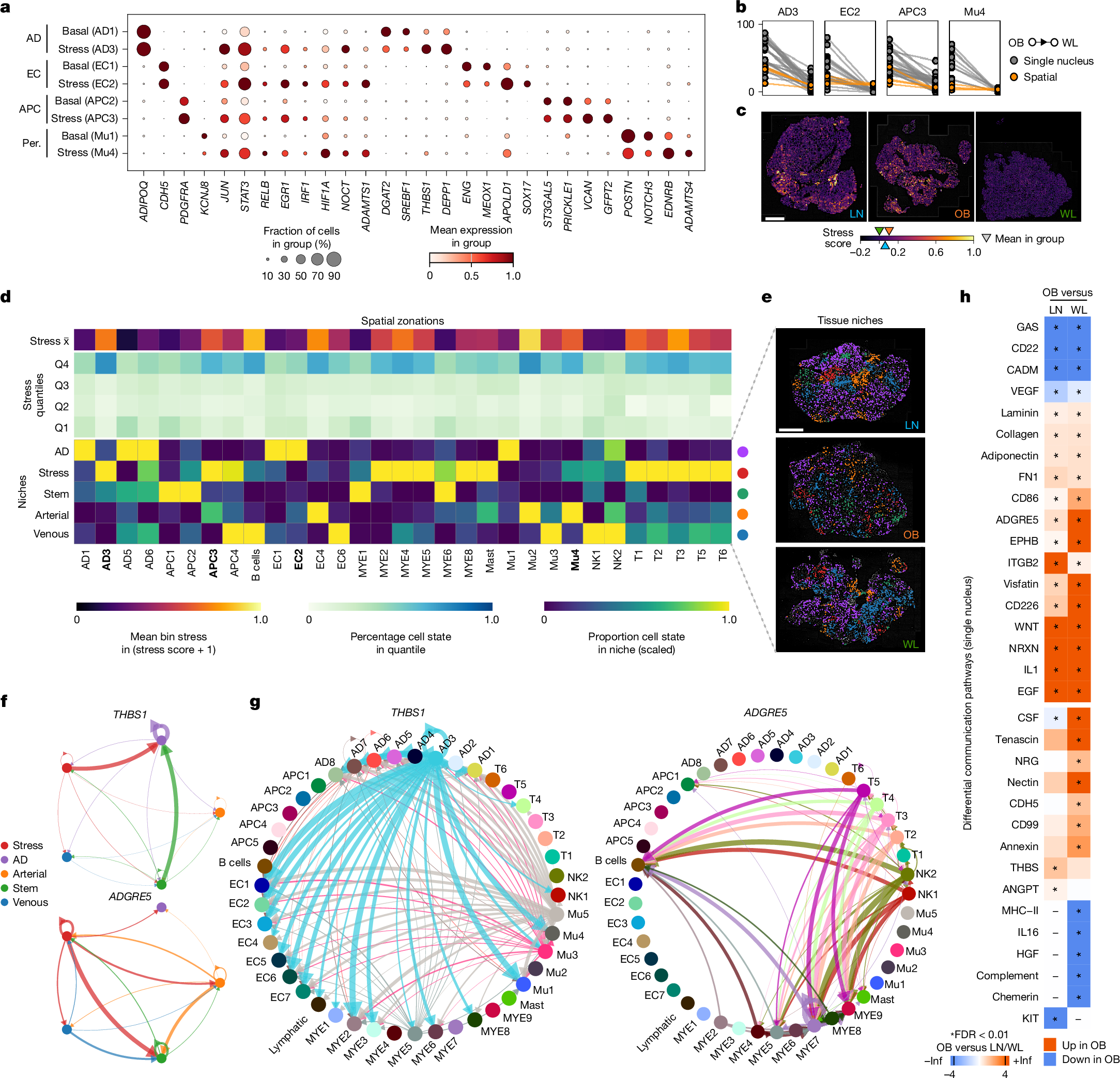
图4 | 应激细胞形成一个空间龛,并富集于应激相关的信号通路

扩展数据图4 | 肥胖和WL中成熟脂肪细胞分子异质性及其调控
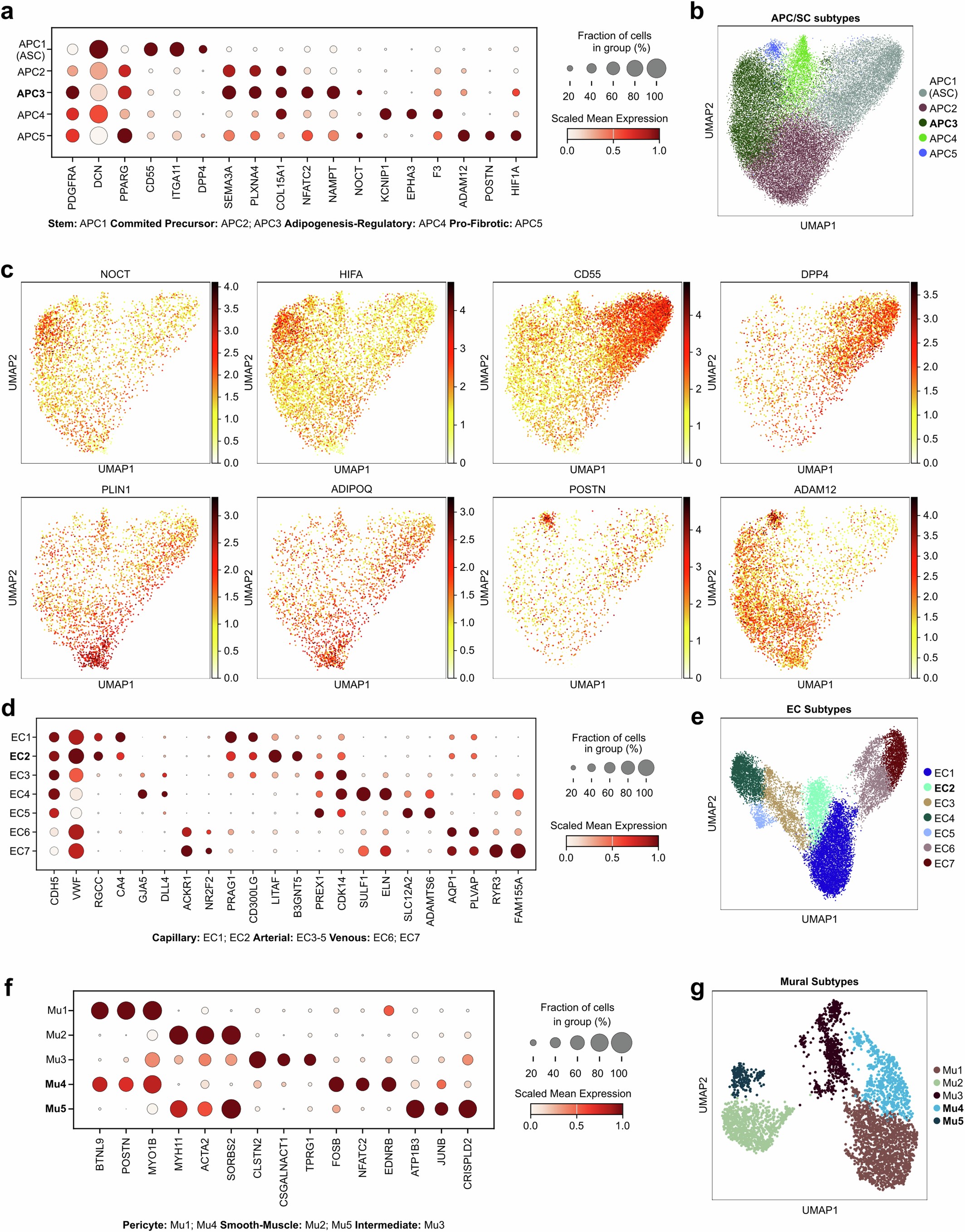
扩展数据图5 | 肥胖和WL的前体细胞和血管细胞表型和适应
5 .多细胞应激的逆转
脂肪祖细胞(Adipocyte progenitor cells,APCs)不仅负责再生成熟的脂肪细胞,也维持着脂肪组织基质稳态,而肥胖症可能会损害这些重要的功能。研究人员首先将APCs分为:“多能”DPP4-CD55-hi祖细胞(ASC/APC1);“定向”的前脂肪细胞(APC2和APC3,表达典型分化基因);脂肪生成调控细胞(APC4、KCNIP-hi和CD142/F3-hi);以及促纤维化前体(APC5、ADAM12-hi和POSTN-hi)(扩展数据图5a-c和补充表2)。肥胖脂肪组织中,APC3表现出与成熟脂肪细胞相似的应激特征,并且高表达抑制分化的NOCT(图4a和扩展数据图5c)。APC2选择性表达脂肪细胞成熟后期基因(扩展数据图5c)。应激(APC3)和促纤维化(APC5)亚群均高表达低氧诱导因子1A(hypoxia-inducible factor 1A,HIF1A)(扩展数据图5a和6a,c),HIF1A能直接抑制PPARγ的转录,此外,它还能通过PDGFR-ERK信号传导驱动PPARγ S112磷酸化,进而导致PPARγ活性下降,最终导致脂肪分化抑制。然而在WL情况下,这两个亚群数量显著减少(图4b和扩展数据图6a,b)。相应地,WL下调了缺氧,促纤维化(TGFβ)和抗脂肪生成(WNT)基因(扩展数据图6c和补充表7和8)。因此,WL可减轻缺氧诱导的分化能力下降,以及某些人类APC亚群中的促纤维化信号。
血管网络的协调生长对AT的健康扩张至关重要。研究人员在血管细胞亚群中也观察到了类似于其他组织类型中的内皮(动脉、毛细血管和静脉)和壁细胞(平滑肌和周细胞)分区(扩展数据图5d-g和补充表2)。与成熟脂肪细胞和APCs一样,肥胖同样诱导了“应激型”毛细血管内皮和壁细胞(图4a,扩展数据图6a,b)。处于应激状态的内皮细胞高表达APOLD1和SNAI1,表明了潜在的病理性新生血管的形成和内皮--间充质的转化过程(图4a和附表4)。处于应激状态的壁细胞高表达 ADAMTS1 ,这是一种与周细胞脱落、纤维化转变和毛细血管稀疏有关的抗血管生成蛋白(图4a)。在不同的单核和空间数据集中,WL明显减少了应激状态血管细胞数量和相关标志物的表达,这意味着病理转化是可逆的(图4c和扩展数据图6a)。
所有应激细胞状态都上调了一个共同的基因特征(涵盖缺氧、机械张力、氧化应激、DNA损伤等基因)(扩展数据图6d-e和补充表12)。多细胞应激虽然在肥胖中最高,但也是瘦组织的一个特征,它随着年龄的增长和代谢功能障碍而增加(图4c和扩展数据图6f,g)。基因和通路分析揭示了多细胞应激的潜在介质(缺氧、机械和氧化应激、Gp130介导的细胞因子、DNA损伤和细胞周期停滞)(扩展数据图7a,b)。体外诱导DNA损伤(使用Etoposide)重现了体内对应激标志蛋白的影响,并损害了ASPC的分化能力(扩展数据图7c-e)。WL导致多细胞应激基因显著减少,从整体上强调了多细胞应激途径在组织损伤和修复中的重要性(扩展数据图6c)。
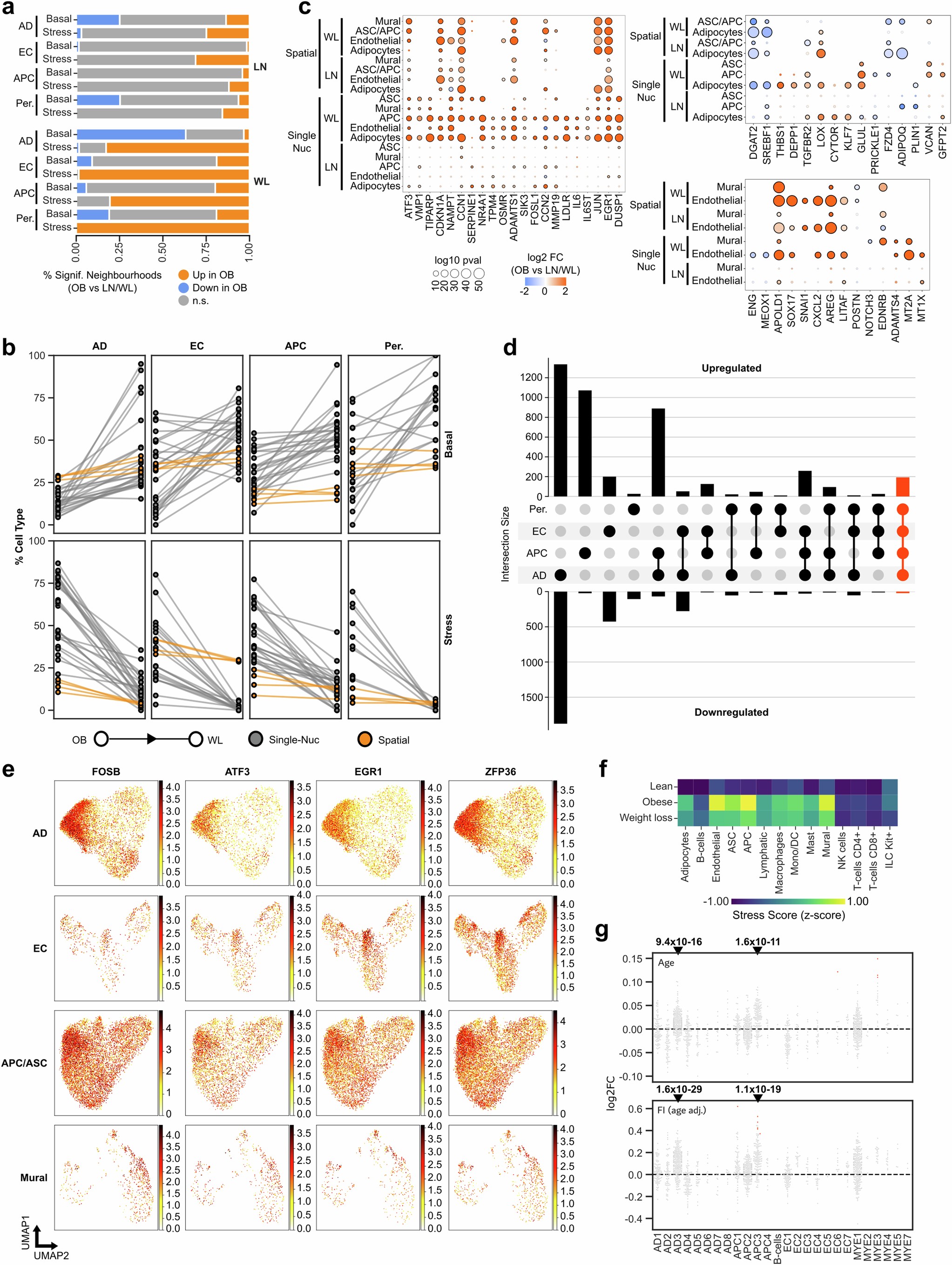
扩展数据图6 | 应激信号在易感细胞类型之间是保守的
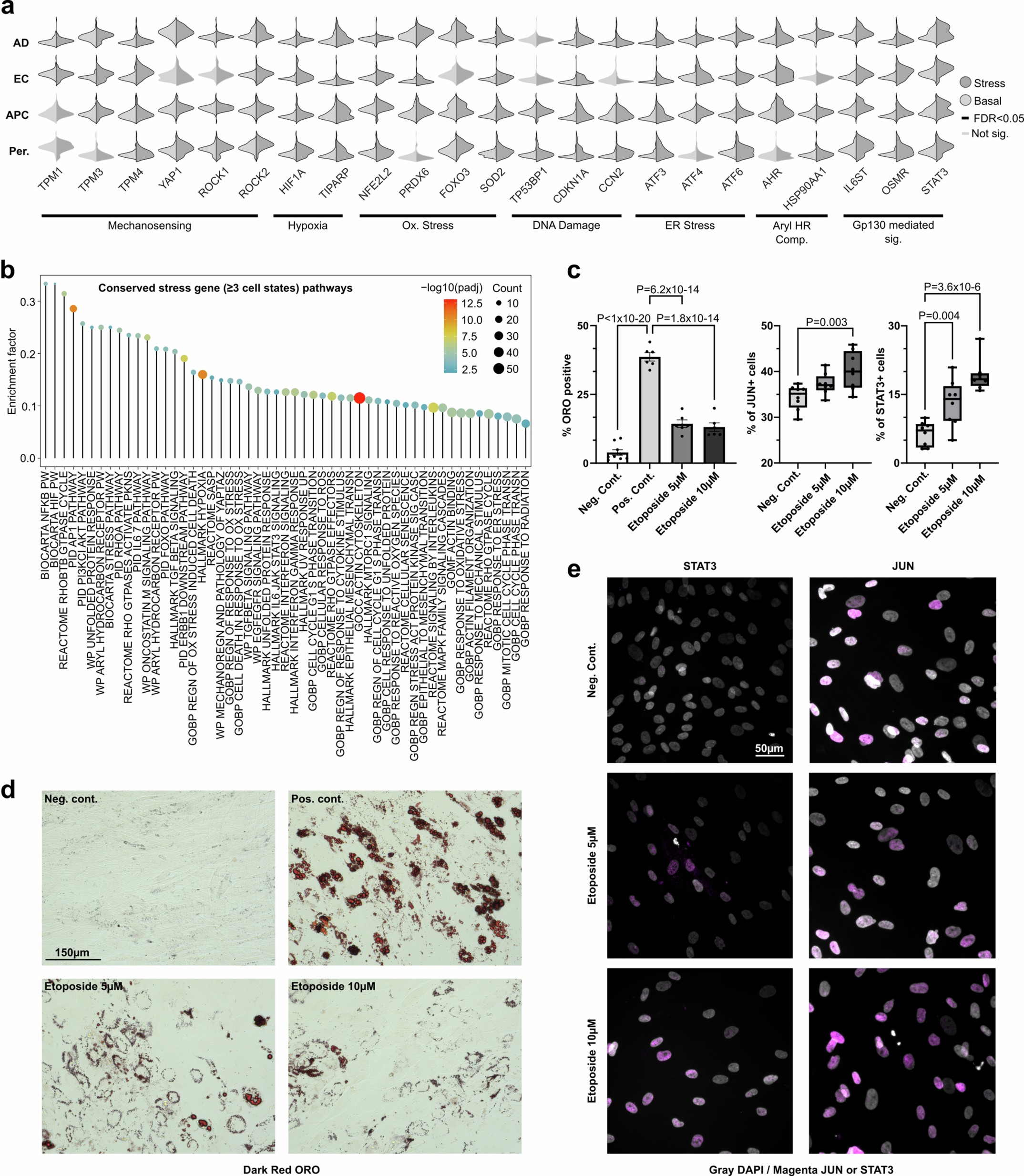
扩展数据图7 | 脂肪组织中细胞应激的调控
6 .改变组织生态位和细胞串扰
研究人员利用空间数据集来研究组织中应激细胞的定位和它的影响,他们量化了低应激区和高应激区的细胞组成(50µm bins;图4d和扩展数据图8a,b)。除了应激毛细血管(EC2)分布遍及整个组织外,其他应激细胞通常富集在高应激区。研究人员还发现,应激区域与免疫细胞共定位,并且与动脉ECs(EC4)存在联系(图4d)。
虽然这个方法将单个细胞的状态定位到了应激区,但它没有深入分析这些细胞是如何在这些微环境区室中组织起来的。为了评估这一点,研究人员使用空间分辨率邻域富集算法(在300μm范围内捕获脂肪细胞),他们进一步定义了5个不同的细胞群落,分别命名为动脉、静脉、脂肪细胞、干细胞和应激生态位(图4d-e和扩展数据图8c)。研究人员发现没有任何一种细胞类型是某一生态位专属的,这表明观察到的细胞分布模式反映的是组织间的梯度变化。干细胞生态位富集到多能性ASC/APC1和同源TRMs。应激生态位富集了AD3、APC3、LAMs、其他先天性(cMono和cDC2)和适应性(T细胞)免疫细胞,表明这些状态与应激诱导和/或反应有关。动脉内皮细胞形成了自己的生态位,与应激前体脂肪细胞(APC3)和应激壁细胞(Mu4)结合在一起。直接的细胞-细胞共定位发现免疫细胞接近大静脉血管和LAMs,这可能反映了它向CLS的外渗和迁移(扩展数据图8d)。
组织分区的确定使研究人员能够研究细胞内和细胞间的信号模式。在空间转录组数据集中进行的配体-受体互作分析揭示了一个复杂的通信网络。脂肪因子和神经营养因子富集在脂肪细胞生态位(ADIPOQ、LEP和NRXN3)(扩展数据图8e-f)。典型的WNT和ECM组分(FN1、胶原蛋白和层粘连蛋白)是干细胞生态位的主要组分(扩展数据图8e)。应激和动脉生态位富含促炎性细胞因子(CXCL2、CCL2和IL6)和推测的应激信号分子(TGFB1、AREG、NAMPT、THBS1)(图4f,扩展数据图8e-f)。研究人员在更大的单核数据集中进行的平行细胞间通讯分析,将不同的生态位信号与源细胞和目标细胞以及疾病病理生物学联系起来(图4g-h和扩展数据图8g-h)。例如,THBS1(应激AD3),ADGRE5(泛免疫)和NAMPT(多细胞),它们是胰岛素抵抗,免疫糖酵解代谢和炎症的新诱因,在肥胖中都被放大,并被WL逆转(图4h和扩展数据图8h)。这表明,应激生态位中高度富集了同时参与病理性组织重塑和恢复性组织重塑的信号分子。
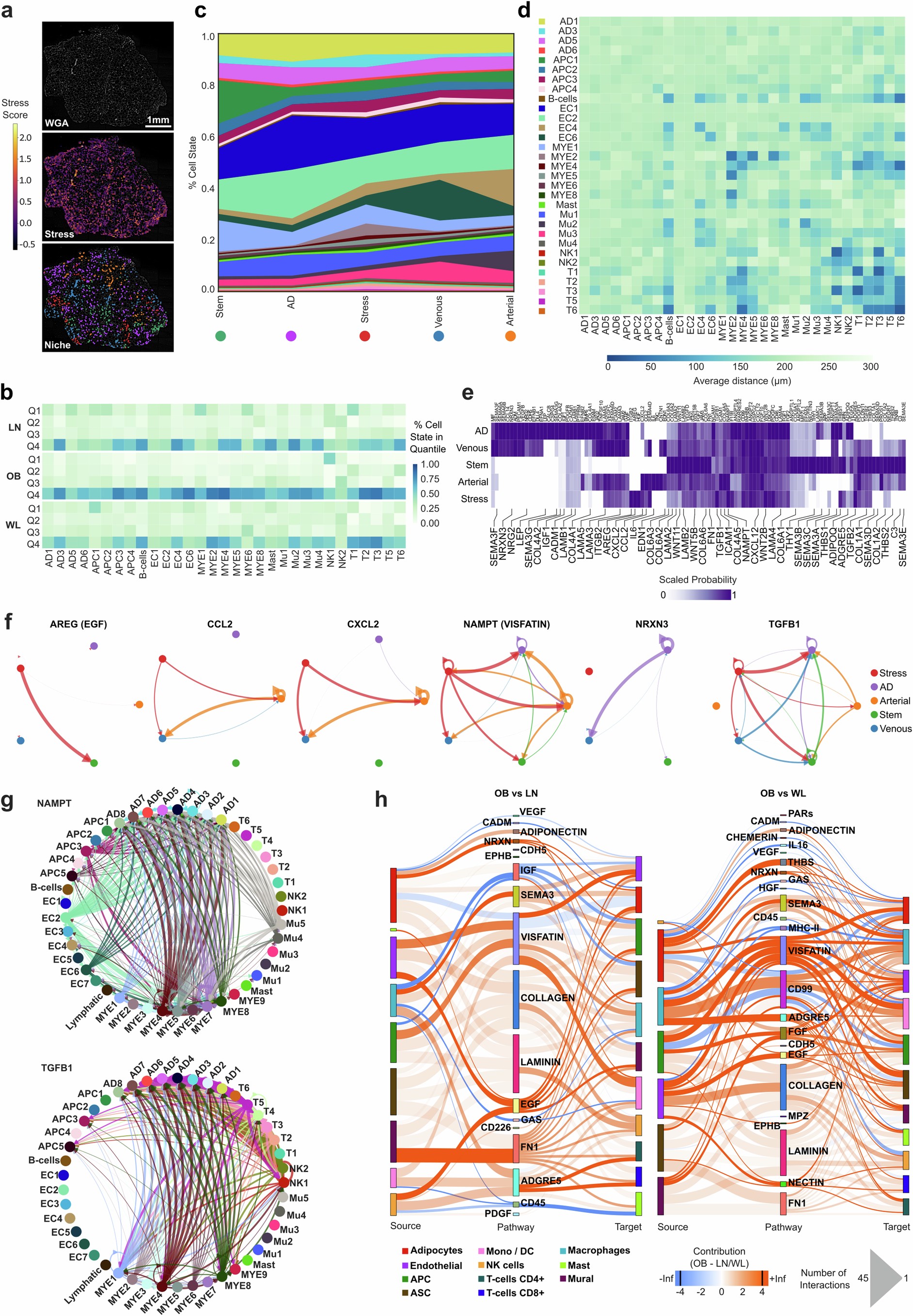
扩展数据图8 | 组织生态位和全组织交流模式
7.抑制衰老
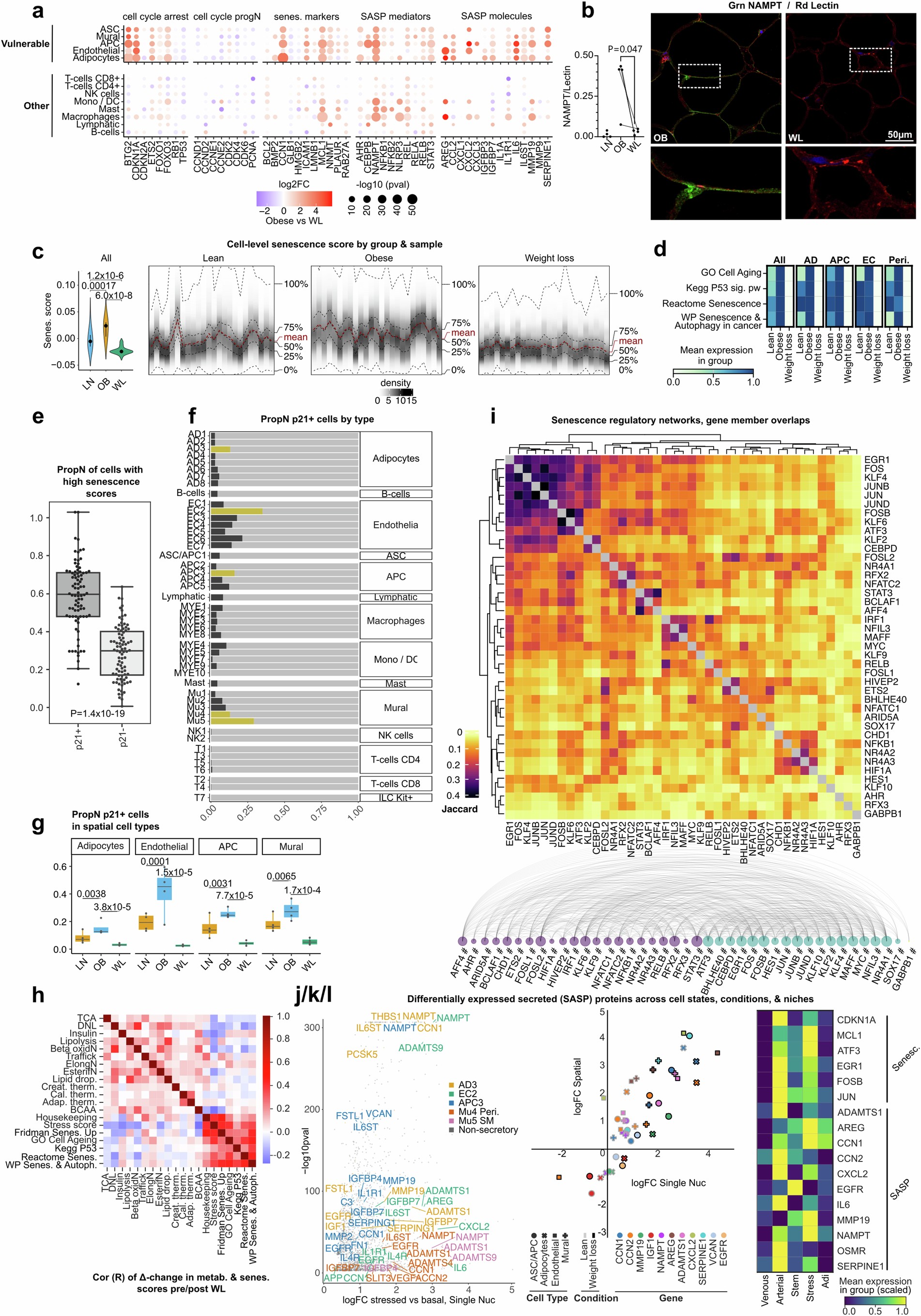
通过差异表达分析确定了AT重塑途径,WL可广泛逆转肥胖对基因调控的影响(扩展数据图 9a,b)。许多与WL相关的最显著的转录变化在不同细胞类型间是保守的,表明这些基因及其调控的通路可能代表了关键的WL机制(扩展数据图9c,d)。WL在多种细胞类型中改变的基因都显示出系统性下调。这些下调的基因是AT功能障碍的标志性通路:炎症(TNFA和IFNG);缺氧;纤维化;免疫细胞的募集和激活;氧化应激(扩展数据图9e)。WL还导致细胞周期停滞基因的下调,共同表明逆转细胞衰老可能是WL改善炎症和代谢的基础(扩展数据图9e)。
为验证此假说,研究人员检测并证实了WL系统性地抑制了衰老细胞标志物的表达(图5a和扩展数据图10a)。在多种类型的细胞中,WL导致衰老过程中主要细胞周期抑制因子CDKN1A(p21)的下调,并恢复被p21抑制的细胞周期进程基因的表达。相应地,WL显著降低了主要衰老标记物的表达和无偏向衰老评分(图5a和扩展数据图10a-d)。研究人员发现,具有衰老细胞转录特征的p21阳性细胞在应激的脂肪细胞、前体细胞和血管细胞状态中最为普遍,这些共同的应激模式反映了细胞的易感性和向衰老转化的趋势(扩展数据图10e-f)。瘦人的AT也含有大量p21阳性细胞(尽管数量明显较少)(图5b)。相比之下,WL几乎完全消除了组织中的p21阳性细胞(图5b),该结果通过空间转录组原位验证(扩展数据图10g)和蛋白定量分析(图5c)得到证实。衰老程序的抑制与脂肪细胞生物能的增强同步发生,表明二者存在机制耦合(扩展数据图10h)。因此,研究人员确定人类 WL 具有以前未曾描述过的强效抗衰老作用。
全组织基因调控网络分析显示,在应激的衰老细胞中有一个紧密保守的转录关系网,该关系网在肥胖时增加,而在WL时减少(图 5d)。鉴定出的TFs可分为几类(图5e和扩展数据图10i): AP-1超家族为衰老基因组提供原动力;激活炎症和衰老相关分泌表型(senescence-associated secretory phenotype ,SASP)的特征性信号依赖的TFs;Krüppel-Like TFs 与细胞周期停滞有关;控制纤毛生成的TFs(RFX2/RFX3),它是一种定向的衰老调节因子;由DNA损伤和氧化应激诱导的孤儿核受体TFs,它们是衰老的关键触发因子;以及多种以前与衰老无关的候选TFs。单个TFs表现出自我调节作用,并共享多个靶基因,包括CDKN1A,表明这些TFs可能协同增强细胞应激、衰老、SASP释放、炎症和组织损伤的退行性循环(图5e和扩展数据图10i)。而这种转录级联反应被WL关闭。
由于AT SASP在强化衰老方面的重要性,研究人员试图通过系统地比较应激(高度衰老)和基础细胞状态下分泌蛋白的表达,来进一步确定AT SASP的特征。这揭示了衰老、组织损伤和代谢功能障碍等多种介质的变化,其中包括与多细胞应激以及细胞内和细胞间通讯相关的信号肽(AREG, ADAMTS1, OSMR, IL6ST 和CXCL2)(扩展数据图10j和补充表12)。研究人员推测的SASP成分在组织中系统性重现,并定位于应激和动脉壁生态位(图5f和扩展数据图10k,I)。衰老细胞强烈上调NAMPT,是SASP的细胞内驱动因子(通过NAD补救途径中的酶活性),也是一种细胞外脂肪细胞因子(黏蛋白),具有多效性、环境依赖性和主要促炎作用(图5a和扩展数据图10j)。NAMPT的表达在肥胖巨噬细胞和炎症LAMs中也有同样的富集,这与其在炎症小体激活和免疫招募中的作用一致(图2b,f)。组织水平的蛋白分析证实,NAMPT的丰度在肥胖时会增加,而在WL时会显著降低,这共同表明NAMPT可能是AT SASP的驱动因素(扩展数据图10b)。
总之,这些分析揭示了退行性AT衰老周期的多种细胞内及细胞外介质,并证实逆转AT衰老是WL改善代谢健康效益的关键决定因素。
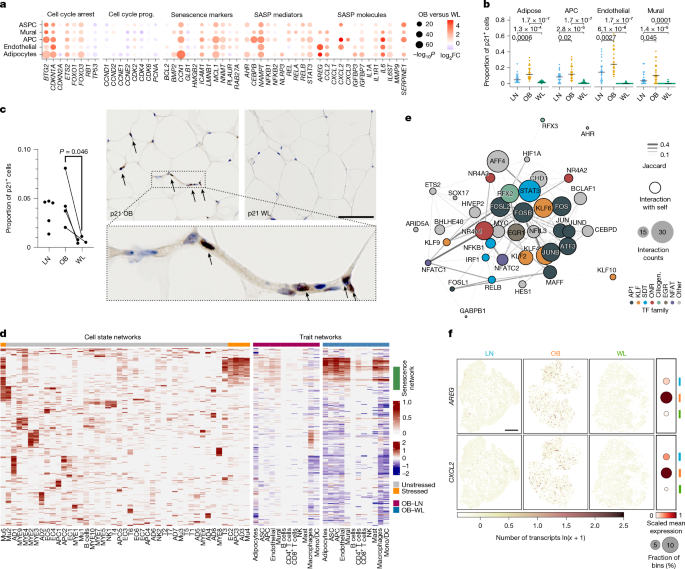
图5 | WL能有效地逆转衰老及其介质
扩展数据图9 | 系统的差异基因表达和通路分析在人类肥胖和WL跨越脂肪组织细胞类型的全谱
扩展数据图10 | 人脂肪组织细胞类型的衰老易感性和调控途径及WL的缓解作用
总结
减肥显著改善肥胖症患者的代谢和心血管健康。脂肪组织(AT)的重塑是这些不同的重要临床现象的核心所在。然而,令人惊讶的是,人们对其潜在机制的了解却很少,这阻碍了治疗的进展。在此,研究人员报告了一个空间分辨单核图谱(包括来自70人的171,247个细胞),该图谱研究了在肥胖和治疗性减肥过程中重塑人类AT从而重塑代谢健康的细胞类型、分子事件和调控因子。他们在代谢细胞、前体细胞和血管细胞中发现了对衰老的选择易感性,并揭示了减肥可以有效地逆转衰老。他们确定了可能驱动衰老、组织损伤和代谢功能障碍退行性循环的基因调控机制和组织信号。研究人员发现,减肥会减少脂肪细胞肥大和生物力学约束途径,激活了全局代谢通量和生物能基质循环,这可能介导了代谢健康的系统性改善。在免疫区,他们证明减肥抑制肥胖诱导的巨噬细胞浸润,但并不能完全逆转其激活,从而使这些细胞有可能引发潜在的体重反弹并加重代谢功能障碍。在整个过程中,他们将细胞映射到组织壁生态位,以了解组织损伤和恢复的集体决定因素。总的来说,他们补充的单核和空间数据集为了解肥胖AT功能障碍及其通过减肥逆转的基础提供了前所未有的见解,是进行机制和治疗探索的关键资源。
原文链接:https://www.nature.com/articles/s41586-025-09233-2IF: 50.5 Q1
转载本文请联系原作者获取授权,同时请注明本文来自徐凌燕科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3483272-1503661.html?mobile=1
收藏