博文
统计基因、基因组序列长度、GC含量和GC-skew
|
Seqkit2 是 Seqkit 的新版本,是一个专门用于处理和分析生物序列数据的软件工具。它支持多种序列数据格式,包括FASTA、FASTQ等,并提供了一系列有用的功能,如数据处理、过滤、统计、格式转换等,是生物信息学领域中常用的工具之一。以下是使用seqkit统计基因、基因组序列长度、GC含量和GC-skew 。
fx2tab子命令:将FASTA/Q转换为表格格式,可用于统计序列的信息
# Usage: seqkit fx2tab [flags]
# flags:
-a, --alphabet: 打印字母表字母。
-q, --avg-qual: 打印读取的平均质量。
-B, --base-content strings: 打印碱基含量。支持多个值,例如 -B AT -B N。
-C, --base-count strings: 打印碱基计数。支持多个值,例如 -C AT -C N。
-I, --case-sensitive: 计算区分大小写的碱基含量/序列哈希。
-g, --gc: 打印 GC 含量。
-G, --gc-skew: 打印 GC-Skew。
-H, --header-line: 打印标题行。
-l, --length: 打印序列长度。
-n, --name: 仅打印名称(不包括序列和质量)。
-Q, --no-qual: 即使对于 FASTQ 文件,也仅输出两列。
-i, --only-id: 打印ID而不是完整标题。
-b, --qual-ascii-base int: ASCII基数,Phred+33为33(默认为33)。
-s, --seq-hash: 打印序列的哈希(MD5)。
# 帮助信息查看
seqkit fx2tab -h
# 打印序列长度、GC含量,并输出前10行
seqkit fx2tab viral.1.1.genomic.fna.gz -l -g -n -i -H | head

# 打印序列长度、GC含量,将统计结果导出为制表符分隔文件
seqkit fx2tab viral.1.1.genomic.fna.gz -l -g -n -i -H > viral.1.1.genomic.fna_length_GC.tsv

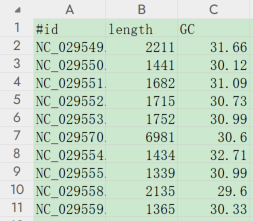
# -l 统计序列长度
# -g 统计平均GC含量
# -i 只打印名称(不打印序列)
# -H 打印标题行
我们将持续分享微生物组学研究和生信分析的专业技能资料。相关课程请于浏览器搜索“密码子学院”。课程问题或个性化分析需求,请联系小唯(微信号:winnerbio01)。
https://wap.sciencenet.cn/blog-3447233-1491727.html
上一篇:RNA/DNA序列相互转换
下一篇:fastq格式文件转换成fasta文件