
 精选
精选
研究背景
人工固碳是现代化学领域在可持续能源和有效减排二氧化碳方面面临的最具挑战性的研究课题之一。将太阳能驱动的光阳极与暗阴极耦合是一种具有应用前景的策略,从而实现氧气的生成和二氧化碳还原,为模拟自然光合作用提供了一种切实可行的解决方案。因此,如何构建具有高活性的析氧光阳极以及高选择性的CO₂还原阴极成为光电催化固碳的关键所在。

Highly Active Oxygen Evolution Integrating with Highly Selective CO₂-to-CO Reduction
Chaowei Wang, Laihong Geng, Yingpu Bi*
Nano-Micro Letters (2025)17: 184
https://doi.org/10.1007/s40820-025-01688-2
本文亮点
1. 本文提出了一种简单可行的策略,可以合理调节光阳极和阴极表面活性位点的配位环境和电子结构。
2. 通过降低BiVO₄光阳极表面修饰的FeNi催化剂的配位数,在AM 1.5G的模拟太阳光以及1.23 VRHE下达到了6.51 mA cm⁻2的优异的水氧化活性。
3. 将单原子钴锚定在富氮的碳材料上,以增加Co-N配位数,显著的促进了CO₂的吸附和活化,实现高选择性的CO₂还原至CO。
内容简介
人工固碳是实现碳循环和环境治理的一条有前景的途径。然而,析氧反应动力学缓慢和二氧化碳还原选择性差严重限制了太阳能到化学燃料的整体转化效率。因此,中国科学院兰州化学物理研究所毕迎普等人提出了一种简单可行的策略,可以合理调节光阳极和阴极表面活性位点的配位环境和电子结构。其中缺陷工程用于降低修饰在BiVO₄光阳极表面的超薄FeNi催化剂的配位数,从而达到了6.51 mA cm⁻2(1.23 VRHE,AM 1.5G)的优异水氧化活性。此外,单原子酞菁钴(II)锚定在富氮碳基底上以增加Co-N配位数,显著促进了CO₂的吸附和活化,从而高选择性生成CO。将BiVO₄/NiFe-Ov光阳极和阴极CoPc-NC耦合进行光电催化CO₂还原,CO生成速率达到了109.4 μmol cm⁻2 h⁻1,且CO的法拉第效率>90%,将此PEC CO₂还原体系进一步与吸收500 nm波长以上太阳光的硅太阳能电池集成,最终太阳能到化学品的转换效率高达5.41 %。
图文导读
I BiVO₄/NiFe-Ov光阳极的表征
通过简单的Ar等离子体处理制备了低氧配位NiFe催化剂修饰的BiVO₄光阳极。BiVO₄光阳极的扫描电子显微镜图像说明其直径为200-300 nm的蠕虫状纳米多孔形貌以及相对光滑的表面。高分辨率透射电子显微镜(HR-TEM)图像清楚地表明,0.293 nm的晶格间距对应于单斜BiVO₄的(040)晶面。值得注意的是,在修饰NiFe-Ov催化剂后,BiVO₄光阳极的表面由光滑转变为粗糙的絮状结构(如图1 A)。图1B是BiVO₄/NiFe-Ov光阳极的横截面SEM图像,光阳极薄膜的平均厚度约为1 μm。HR-TEM图像(图1D)清楚地显示,厚度约为5 nm的NiFe-Ov催化剂的非晶纳米层均匀地覆盖在BiVO₄纳米晶体的表面。此外,图1C显示了EDS元素映射分析,表明NiFe催化剂位于BiVO₄表面。图1E显示了原始BiVO₄和BiVO₄/NiFe-Ov光阳极的X射线衍射(XRD)图。与原始BiVO₄样品相比,由于NiFe-Ov催化剂的非晶态结构和超薄厚度,衍射峰没有明显变化。此外,X射线光电子能谱(XPS)高分辨率O 1s光谱清楚地表明,与原始的BiVO₄相比,BiVO₄/NiFe-Ov光阳极上的氧空位显著增加(图1F),证实了NiFe-Ov催化剂上形成了丰富的氧空位。此外,与原始BiVO₄相比,BiVO₄/NiFe-Ov的O 1s峰向低结合能(~0.08 eV)区域移动,这归因于氧空位引起的电子富集。
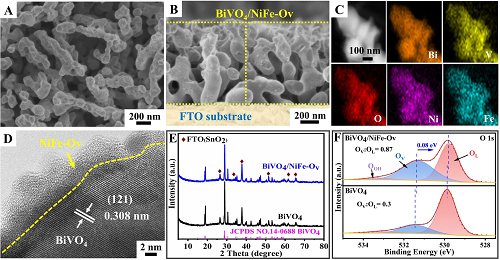
图1. A BiVO₄/NiFe-Ov光阳极的SEM图像、B Cross-SEM图像、C EDS图像和D HR-TEM图像;BiVO₄和BiVO₄/NiFe-Ov光阳极的E XRD图谱和F XPS高分辨率O 1s光谱。
II BiVO₄/NiFe-Ov光阳极的光电化学性能
在0.5 M K₃BO₃(pH=9.5)电解质中,在AM 1.5G光照(100 mW cm⁻2)下,对原始BiVO₄和BiVO₄/NiFe-Ov光阳极的PEC水氧化活性进行了研究。如图2A所示,在1.23 VRHE下,相比于原始BiVO₄电流密度为1.55 mA·cm⁻2,BiVO₄/NiFe-Ov光阳极的光电流密度可以显著增加到6.51 mA·cm⁻2,表明光阳极/电解质界面的析氧活性明显改善。如图2B所示,BiVO₄/NiFe-Ov光阳极的ABPE值在0.72 VRHE下高达1.98%,远高于原始BiVO₄的ABPE值(在0.94 VRHE下为0.25%)。此外,图2C显示了原始BiVO₄和BiVO₄/NiFe-Ov光阳极的入射光子到电流转换效率(IPCE)。与原始BiVO₄相比,BiVO₄/NiFe-Ov光阳极在360 nm波长处的最大值为93.5%。此外,还进行了电化学阻抗谱(EIS)测试,以阐明界面电荷分离和转移(图2D)。根据Nyquist图和拟合结果,BiVO₄/NiFe-Ov和原始BiVO₄光阳极的电阻值分别为76.1和665.1 Ω,表明NiFe-Ov催化剂在加速界面电荷转移方面具有较好的能力。此外,PEC水氧化稳定性在1.23 VRHE下进行,如图2E所示。可以明显发现,在修饰NiFe-Ov催化剂后,BiVO₄光阳极的光电流密度在6小时的测试过程中可以很好地保持稳定,这表明NiFe-Ov对抑制V⁵⁺从BiVO₄晶格中溶解具有建设性作用。此外,通过气相色谱法(GC)测定了BiVO₄/NiFe-Ov光阳极上PEC水分解产生的氢气和氧气量。如图2F所示,H₂和O₂的产量随反应时间的延长呈线性增加。经过3小时的光照后,H₂和O₂产物的量分别可达362.9(H₂)和180.8 μmol(O₂),BiVO₄/NiFe-Ov光阳极的平均法拉第效率为93.2%,进一步证实了其突出的水氧化能力。因此,合理降低FeNi基OER催化剂的配位数可以显著提高PEC水氧化性能。
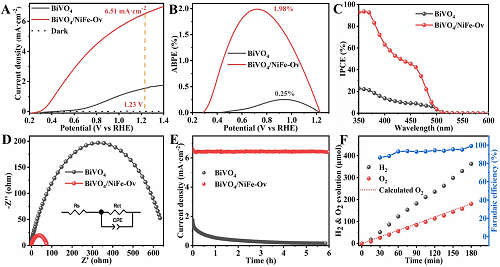
图2. A BiVO₄和BiVO₄/NiFe-Ov光阳极在0.75 VRHE下的LSV(扫描速率为10 mV s⁻1)、B ABPE、C IPCE、D EIS图,以及在1.23 VRHE的E 稳定性和F H₂和O₂析出测试。
III CoPc-NC阴极的表征
为了实现高效的PEC CO₂还原,高活性的光阳极须与高选择性的阴极催化剂结合,以确保CO₂的吸附和活化。在此,我们选择了在富氮的碳基底上锚定的单原子CoPc作为阴极催化剂,与上述BiVO₄/NiFe-Ov光阳极耦合。SEM和TEM图像清楚地显示,所获得的CoPc-NC催化剂具有均匀的菱形十二面体形貌,平均粒径约为250 nm(图3A-B),且没有发现CoPc簇的明显聚集。此外,EDS元素映射图像(图3C)显示,在整个样品区域可以很好地检测到Co、N和C信号,进一步证实了CoPc在NC基底上的均匀分散。球差透射电子显微镜(HAADF-STEM)图像(图3D)证实,Co单原子均匀地锚定在N-C基底上。此外,X射线吸收结构(XAS)光谱也被应用于探究化学结构和配位环境的细节。如图3E所示,CoPc-NC的EXAFS小波变换光谱表明,WT轮廓图的最大强度位于~4.3Å⁻1,对应于~7.2Å⁻1的Co-N键,而不是Co-Co键,证实了富氮碳基底上Co单原子的形成。更具体地说,EXAFS分析的傅里叶变换(图3F)清楚地表明,在~1.47Å和1.51Å处的峰值可以分别归因于CoPc和CoPc-NC的Co-N键。此外,Co-K边XANES光谱表明,与CoPc相比,CoPc-NC的光谱向更高的能量偏移。拟合结果表明,CoPc-NC催化剂中Co单原子的配位环境应该是Co-N₅,这可以进一步得到XPS结果的支持。与原始CoPc样品相比,CoPc-NC催化剂的Co 2p峰向低结合能区域移动,表明电子由NC基底注入单原子Co位点。
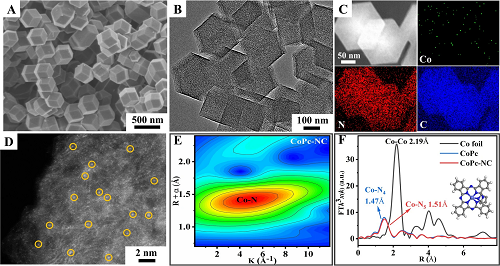
图3. A CoPc-NC的SEM图像、B TEM图像、C EDS映射图像、D HAADF-STEM图像和E EXAFS小波变换光谱;F Co箔、CoPc和CoPc-NC的EXAFS分析。
IV 光电催化CO₂还原性能
在H型电池中,以CO₂饱和的0.5 M KHCO₃为电解质,研究了与BiVO₄/NiFe-Ov光阳极集成的CoPc-NC阴极的PEC CO₂还原性能。如图4A所示,在1.23 VRHE下照射BiVO₄/NiFe-Ov光阳极1小时后,CoPc-NC阴极室产生的CO和H₂量分别增加到109.4和11.3 μmol·cm⁻2。然而,当从BiVO₄/NiFe-Ov光阳极移除光照时,在CoPc-NC阴极上没有检测到任何气体产生,这表明光阳极上PEC水氧化释放电子/质子对于在阴极上驱动CO₂还原反应生成CO至关重要。此外,同位素标记实验是使用13CO₂作为碳源进行的,如图4B所示。在该PEC CO₂还原系统中明显检测到m/z=29处的13CO峰,证实CO是由CO₂而不是其他杂质产生。值得注意的是,通过改变施加电压可以方便地控制CO:H₂的比例(3.5~9.7)(图4C),当施加电势大于0.8 VRHE时,所有电压下的CO法拉第效率都高于85%(图4D)。此外,通过循环实验评估了该PEC CO₂还原系统的稳定性。如图4E所示,CO的产生量和法拉第效率在五个循环中保持不变,证实了其对二氧化碳还原的出色耐久性。为了进一步验证CoPc-NC阴极对PEC CO₂还原的高活性和选择性,NC、Co-NPs-NC、Co-NC和CoPc阴极催化剂也与BiVO₄/NiFe-Ov光阳极偶联。如图4F所示,上述阴极上的CO产量均远低于CoPc-NC。特别是,CoPc的CO法拉第效率仅达到37.9%,表明CoPc-NCs的优异活性和选择性可归因于CoPc与NC基底较好的键合。
为了进一步阐明CO₂在CoPc-NC阴极表面吸附和活化的细节,图4G所示的原位傅里叶变换红外反射(FTIR)光谱显示,与CoPc样品相比,CoPc-NC催化剂上CO₂的弯曲振动(~667 cm⁻1)峰显著增强,表明CoPc-NCs催化剂由于强大的O-C-O弯曲振动而有利于CO₂的活化。因此,CoPc-NC具有比原始CoPc更强的CO₂吸附和活化能力。此外,还进行了准原位XPS光谱,以探究CO₂吸附和活化过程中的电荷转移过程。CoPc和CoPc-NC在CO₂吸附前后的O1s光谱,以及CO₂吸附的O1s峰可分为C=O(~533.2 eV)和C-O(~531.6 eV)两种氧物种。值得注意的是,CoPc-NC表现出比CoPc更大比例的C-O峰,这表明CoPc-NC催化剂具有更高的CO₂吸附和活化能力。如图4H所示,CO₂吸附后,Co 2p峰可能会向高结合能方向(~0.2 eV)移动,表明电子从单原子Co位点转移到CO₂分子。基于上述分析,我们提出了与BiVO₄/NiFe-Ov光阳极耦合的CoPc-NC阴极催化剂上PEC CO₂还原的可能机理(图4I)。在光照下,光生空穴从BiVO₄体相迁移到NiFe表面进行水氧化,Co单原子位点吸附的CO₂分子与OER过程中提供的质子和电子相互作用形成*COOH和*CO,随后CO分子从CoPc-NC表面解离。
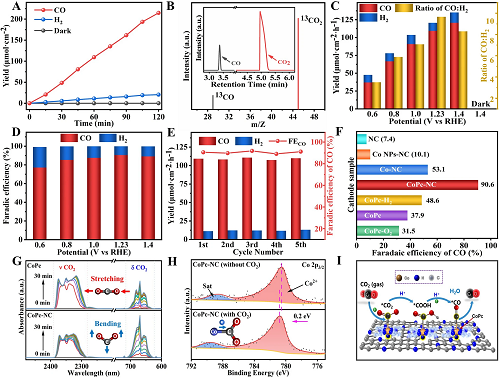
图4. A BiVO₄/NiFe-Ov‖CoPc-NC的CO和H₂析出量为1.23 VRHE;B 13CO₂作为碳源的GC-MS产物分析;不同电位下BiVO₄/NiFe-Ov‖CoPc-NC串联的C CO/H₂产率和D法拉第效率;在1.23 VRHE的循环试验中,E CO/H₂的产量和CO的法拉第效率;F BiVO₄/NiFe-Ov光阳极与不同阴极集成的CO法拉第效率;G CoPc和CoPc-NC对CO₂吸附的原位FTIR光谱;H CoPc-NC在CO₂吸附前后的XPS Co 2p的高分辨率光谱;I BiVO₄/NiFe-Ov‖CoPc-NC耦合系统的PEC CO₂还原路径的机理图。
V 无偏压PV-PEC CO₂还原性能
基于BiVO₄的光阳极只能吸收波长小于500 nm的太阳光谱,用于吸收剩余太阳光(>500 nm)的商用多晶硅太阳能电池与上述PEC装置集成,以构建一个全光谱太阳能驱动的二氧化碳还原装置(图5A-B)。在AM 1.5G光照(100 mW·cm⁻2)下,该PV-PEC CO₂还原系统的光电流密度可达4.1 mA·cm⁻2(图5C)。然而,当BiVO₄光阳极上的光照被移除时,光电流密度迅速降至接近零,在这个PV-PEC反应系统中没有检测到任何气体产物(图5D),证实了PEC CO₂的还原是由BiVO₄/NiFe-Ov光阳极驱动的,而不是太阳能电池。此外,光照1小时后,O₂、CO和H₂产物的量分别可达35.3、62.2和14.7 μmol·cm⁻2,化学计量比为~2:1(CO+H₂/O₂)。如图5E所示,CO和H₂的法拉第效率在10小时的连续光照过程中保持基本不变,表明PV-PEC系统具有优异的稳定性。太阳能到CO和H₂的转换效率分别为4.44%和0.97%,对应于太阳能到燃料(STF)的转换效率为5.41%。此外,图5F显示了近年来报道的光阳极驱动的PEC-CO₂还原的STF转换效率的比较,并且该PV-PEC CO₂还原系统达到了最高的太阳能转换效率。
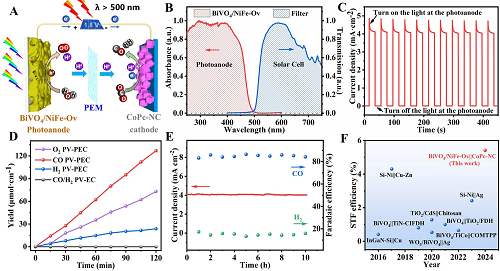
图5. A BiVO₄/NiFe-Ov‖CoPc NC PV-PEC CO₂还原系统示意图;B BiVO₄/NiFe-Ov光阳极和太阳能电池截止滤光片的吸收光谱和透过光谱;C 斩光i-t曲线,D O₂、CO和H₂析出量,E BiVO₄/NiFe-Ov‖CoPc NC PV-PEC CO₂还原系统的稳定性测试;F PEC CO₂还原系统的太阳能到燃料转换效率的比较。
VI 总结
该研究通过合理调节光阳极和阴极表面活性位点配位环境,共同实现了高活性氧生成和高选择性的二氧化碳还原。一个模拟太阳光照射下,低配位的超薄FeNi催化剂修饰的BiVO₄光阳极在1.23 VRHE达到了6.51 mA cm⁻2的优异OER活性。然后与Co-N₅配位的单原子酞菁钴(II)的耦合实现了高效的CO人工光合成,法拉第效率为90.6%。此外,将该PEC cell与光伏电池集成以构建无偏压太阳能驱动的二氧化碳还原装置,太阳能到燃料的转换效率达到了5.41%,且具有出色的稳定性。因此,开发高效的光阳极并与高选择性的二氧化碳还原相结合,是利用太阳能驱动人工光合成高附加值燃料的一种具有前景的策略。
作者简介

毕迎普
本文通讯作者
中国科学院兰州化学物理研究所 研究员
▍主要研究领域
半导体光电催化性能研究;半导体光催化制氢及CO₂还原研究;光生电荷分离迁移动态研究。
▍主要研究成果
毕迎普研究员,博士生导师,精细石油化工中间体国家工程中心副主任。研究工作主要集中于半导体光催化及光电催化纳米材料的结构设计构建及其性能研究,揭示半导体材料的结构与光催化和光电催化性能之间的构效关系,从而构建具有高活性的半导体光催化及光电催化纳米材料。在J. Am. Chem. Soc, Angew. Chem. Int. Ed., Nat. Catal, Nat. Commun., Energy Environ. Sci., Adv. Mater等期刊发表论文150余篇,相关工作被引用16000余次,其中2篇引用超过1000次,H因子为57。先后承担国家自然科学基金重点项目、优秀青年基金、面上项目等10余项,并获得“中国催化新秀奖”,“光催化优秀青年奖”,“国家优秀青年基金”,“中国科学院人才计划考核优秀”等奖励。
▍Email:yingpubi@licp.cas.cn
撰稿:原文作者
编辑:《纳微快报(英文)》编辑部
关于我们







Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2023 JCR IF=31.6,学科排名Q1区前3%,中国科学院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
转载本文请联系原作者获取授权,同时请注明本文来自纳微快报科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3411509-1482707.html?mobile=1
收藏