博文
识别单个肿瘤的癌症驱动基因
||
识别单个肿瘤的癌症驱动基因
虽然治疗癌症的新方法的发展改善了某些癌症类型患者的生存前景,但其他癌症类型的死亡率却急剧上升。例如,在澳大利亚和美国,肝癌、胰腺癌、甲状腺癌和子宫癌的死亡率在过去四十年中稳步上升,预计其死亡率在未来将急剧上升。不出所料,这些癌症通常不能手术,缺乏有效的化疗选择。在肝癌和胰腺癌的病例中,一线化疗只能使预期寿命增加几个月,唯一可能治愈的治疗选择是手术切除,由于合并症,只有不到25%的患者符合晚期手术切除的条件检测与进展。这些癌症通常采用非靶向细胞毒性化疗治疗,包括DNA合成和修复抑制剂、拓扑异构酶抑制剂或广泛性多酪氨酸激酶抑制剂。这些药物的作用几乎没有特异性,它们杀死过度活跃和快速复制的肿瘤细胞的速度比正常的身体细胞要快。
为了使化疗更有针对性地针对癌细胞,人们一直致力于开发分子靶向化疗药物,以靶向具有特定基因改变的细胞。然而,在某些癌症类型中观察到的广泛异质性阻碍了这些治疗方法的广泛适用性。虽然靶向治疗通常对含有相关突变的肿瘤有效,但个体肿瘤的极端异质性限制了其有效性。例如,在肝癌中最常见的6种突变中,没有一种突变出现在超过31%的患者中。Reimand和Bader等人将这种现象定义为长尾假说:癌症突变由一长串频繁突变的基因和一长串不频繁突变的基因组成,这些基因共同构成了大多数驱动突变。
为了解决这个问题,将定向治疗与最有可能产生反应的患者相匹配的个性化方法已成为现代癌症治疗的关键。个性化医疗有许多优点,包括提高药物疗效、减少副作用和降低成本,但一些挑战仍然存在。实现更多以患者为中心的治疗的基本方法是识别生物标志物,预测患者对治疗的反应,然后相应地对患者进行分层。虽然这种方法在某些类型的癌症中取得了成功,但对许多类型的癌症来说却并非如此。例如,对于肝癌,几项主要的多组学研究试图对肿瘤进行分子分类。然而,到目前为止,还没有确定的生物标志物可以可靠地预测对任何治疗的反应,这可能是由于大多数复发性突变的可操作性有限。此外,一些大型临床试验已经开始评估癌症患者真正基于分子生物标志物的治疗选择的可行性,但到目前为止,结果并不令人印象深刻。例如,美国国家癌症研究所(National Cancer Institute)的治疗选择分子分析(NCI-MATCH)迄今为止显示,只有37.6%的患者中存在可操作的突变,并且两组已发表的研究没有显示出阳性结果。同样目的的癌症治疗和优化分子筛选(MOSCATO)试验也发现,只有不到50%的患者携带符合其靶标列表的突变,总体而言,只有7%的患者从试验中受益。
显然,仅仅基于基因变异存在的精确护理是不够的。为了避免这种情况,进一步分析确定驱动基因改变可能会改善这些努力。简而言之,肿瘤通常包含数千个体细胞突变;然而,这些突变中只有一小部分被称为驱动突变,负责驱动癌症的生长,而其余的被称为乘客突变。此外,考虑到驱动突变对于肿瘤生长是不可缺少的,假设在每个肿瘤内亚克隆中都存在一些驱动突变。因此,驱动突变是精确护理的理想目标。
有问题的是,大多数常用的识别驱动基因的方法在队列水平上起作用,例如,MuSiC、MutSigCV、CHASM、HotNet2,通常是突变频率的反映。一旦这些驱动因素被充分表征,它们就会被储存在诸如癌症变异临床解释(CIViC)、癌症基因普查(CGC)、癌症基因网络(NCG)等数据库中。然而,目前尚不清楚这些典型驱动因素是否在所有情况下都是驱动因素,也就是说,如果一个突变导致一个病人的癌症,它可能不一定会导致另一个病人的癌症。事实上,一种真正个性化的癌症治疗方法将受益于能够根据对个体患者造成的影响来识别驱动因素。
最近,Gillman等人对单个肿瘤的癌症驱动基因识别方法进行了系统综述,该综述的重点是驱动程序优先化方法上,该方法试图通过结合基因组和转录组测序数据来识别个性化驱动程序,以评估遗传改变如何影响基因相互作用网络(GIN)中的表达模式。作者们探讨了这些方法的局限性,以及以无重复数据的差异表达分析的emergent方法的形式解决这些局限性的一些潜在解决方案。最后,作者们简要讨论了一些不依赖于转录组学数据的基于机器学习的替代方法。
全文将癌症驱动基因识别方法分为两大类:网络驱动方法(图1)和机器学习方法。网络驱动方法又分为使用外部参考网络的网络驱动方法和使用De-Novo网络的网络驱动方法。
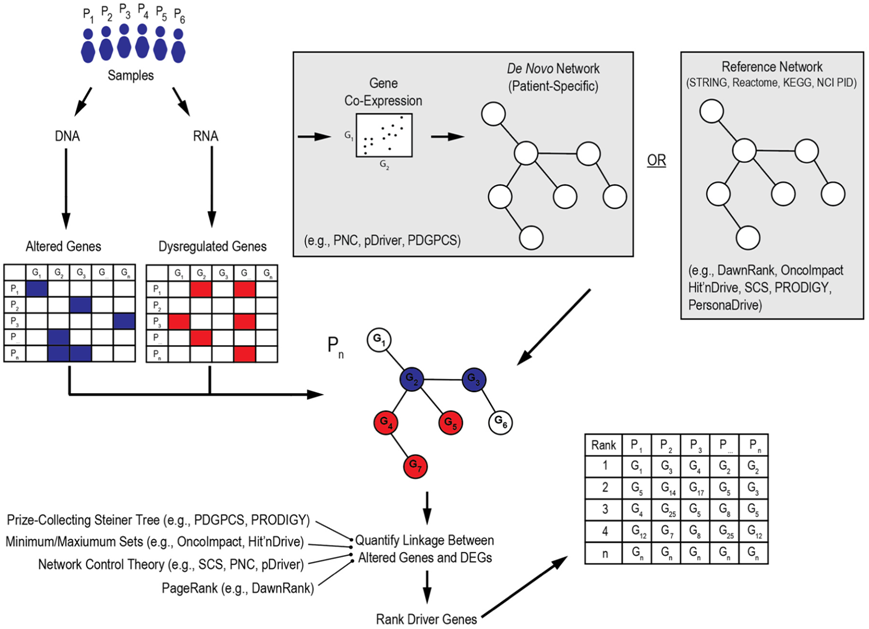
图1 基于网络的驱动基因识别概述。考虑到驱动突变是产生癌症表型的原因,人们假设这种表型一定是基因转录活性改变的结果。因此,通过将突变数据与基因相互作用网络(GIN)上的表达数据相结合,这些算法根据突变如何影响基因表达来量化突变作为驱动因素的可能性。首先,获得每个个体患者的基因组(DNA)和转录组(RNA)测序数据,并利用这些信息确定每个基因的遗传改变(蓝色)和表达失调(红色)。然后将这些改变和失调的基因映射到GIN中,GIN可以来自外部数据库(例如STRING、Reactome、KEGG、NCI PID),也可以从转录组学数据中重新构建。然后,优先排序算法利用各种方法来量化改变基因和差异表达基因之间的联系,并最终根据改变基因对网络的总体影响对其进行排名
近年来,医学领域在在癌症驱动因子优先排序上取得了重大进展,但很少有人关注单个肿瘤的方法。这篇综述讨论了为此目的设计的一些计算方法,其中大多数是基于网络的方法,结合了基因组和转录组测序数据,但也包括只需要基因组数据的机器学习方法。虽然每一种新方法都建立在前一种方法的基础上,并试图解决其固有的局限性,但对于这些方法所预测的驱动因素是否在体外和体内具有生物学意义,以及它们是否可以作为治疗靶点,目前还没有对这些方法进行独立评估。鉴于这些方法在实现个体化治疗癌症患者方面的潜力,进行这样的评估是至关重要的。
参考文献
[1] Gillman R, Field MA, Schmitz U, Karamatic R, Hebbard L. Identifying cancer driver genes in individual tumours. Comput Struct Biotechnol J. 2023 Oct 13;21:5028-5038. doi: 10.1016/j.csbj.2023.10.019.
以往推荐如下:
5. EMT标记物数据库:EMTome
8. RNA与疾病关系数据库:RNADisease v4.0
9. RNA修饰关联的读出、擦除、写入蛋白靶标数据库:RM2Target
13. 利用药物转录组图谱探索中药药理活性成分平台:ITCM
19. 基因组、药物基因组和免疫基因组水平基因集癌症分析平台:GSCA
22. 研究资源识别门户:RRID
24. HMDD 4.0:miRNA-疾病实验验证关系数据库

https://wap.sciencenet.cn/blog-571917-1410009.html
上一篇:利用agoTRIBE检测单细胞转录组内miRNA靶标
下一篇:NetSeekR:RNA转录组时序数据的网络分析管道