博文
听哈佛大神讲怎么做单细胞转录组GSEA分析
||
小剧场:
记得有一天,我正准备兴匆匆的给我的单细胞亚群间的差异基因跑个GO富集分析的时候,我的小老板迈着她那猥琐的步伐悄悄的跑到我身后,愣了一阵儿,说:“小虎子,怎么还跑GO啊,都过时了!现在都跑GSEA!”
我睁开我蒙昧的小眼睛:“老师,啥叫GSEA啊?”
老师愣了一下,“这么简单都不会,自己查查去”。
。。。我。。。。我的老板应该不知道GSEA是什么。。。
GSEA定义
Gene Set Enrichment Analysis (基因集富集分析)用来评估一个预先定义的基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。其输入数据包含两部分,一是已知功能的基因集 (可以是GO注释、MsigDB的注释或其它符合格式的基因集定义),一是表达矩阵,软件会对基因根据其与表型的关联度(可以理解为表达值的变化)从大到小排序,然后判断基因集内每条注释下的基因是否富集于表型相关度排序后基因表的上部或下部,从而判断此基因集内基因的协同变化对表型变化的影响(一文掌握GSEA,超详细教程)。
(The gene sets are defined based on prior biological knowledge, e.g., published information about biochemical pathways or coexpression in previous experiments. The goal of GSEA is to determine whether members of a gene set S tend to occur toward the top (or bottom) of the list L, in which case the gene set is correlated with the phenotypic class distinction.)
这与之前讲述的GO富集分析不同。GO富集分析是先筛选差异基因,再判断差异基因在哪些注释的通路存在富集;这涉及到阈值的设定,存在一定主观性并且只能用于表达变化较大的基因,即我们定义的显著差异基因。而GSEA则不局限于差异基因,从基因集的富集角度出发,理论上更容易囊括细微但协调性的变化对生物通路的影响。
我不知道大家看懂上面那段话没有,我换句话说一遍:GSEA与GO分析的重要区别在于它使用的不是差异基因集而是经排序(p值或者logFC)的全部基因列表。
GSEA在单细胞转录组中的应用

大神!!!!!!!!!!!!!!!!(悄悄说,长的超像我大老板。。。)
按照在哈佛大学FAS信息学scRNAseq研讨会发表的部分课程,我们已经提前下载该数据public 5k pbmc (Peripheral blood mononuclear cell) dataset from 10x genomics[1] ,并且通过Seurat经典的workflow进行分析运行(参考[2]):
下载数据
wget http://cf.10xgenomics.com/samples/cell-exp/3.0.2/5k_pbmc_v3/5k_pbmc_v3_filtered_feature_bc_matrix.tar.gz tar xvzf 5k_pbmc_v3_filtered_feature_bc_matrix.tar.gz
加载R包
library(tidyverse) library(Seurat)
workflow
# Load the PBMC dataset
pbmc.data <- Read10X(data.dir = "data/pbmc5k/filtered_feature_bc_matrix/")
# Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc5k", min.cells = 3, min.features = 200)
pbmc
#QC
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# Visualize QC metrics as a violin plot
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 5000 & percent.mt < 25)
#Normalization
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
#高变基因选择
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
#标准化
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
#去除MT,重新进行标准化
pbmc <- ScaleData(pbmc, vars.to.regress = "percent.mt")
#PCA
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc), verbose = FALSE)
#聚类
pbmc <- FindNeighbors(pbmc, dims = 1:20)
pbmc <- FindClusters(pbmc, resolution = 0.5)
#可视化
pbmc <- RunUMAP(pbmc, dims = 1:20)
pbmc<- RunTSNE(pbmc, dims = 1:20)
## after we run UMAP and TSNE, there are more entries in the reduction slot
str(pbmc@reductions)
DimPlot(pbmc, reduction = "umap", label = TRUE)
#查找marker基因
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
saveRDS(pbmc, "data/pbmc5k/pbmc_5k_v3.rds")如图所示在处理以后,进行细胞分群后可以分为14个细胞亚群,分别为cluster 0-13。

library(Seurat)
pbmc<- readRDS("data/pbmc5k/pbmc_5k_v3.rds")
# 对于GSEA,需要所有基因的信息
# all 20,000 genes. instead let's try presto which performs a fast Wilcoxon rank sum test
#library(devtools)
#install_github('immunogenomics/presto')
library(presto)
Loading required package: Rcpp
pbmc.genes <- wilcoxauc(pbmc, 'seurat_clusters')
head(pbmc.genes)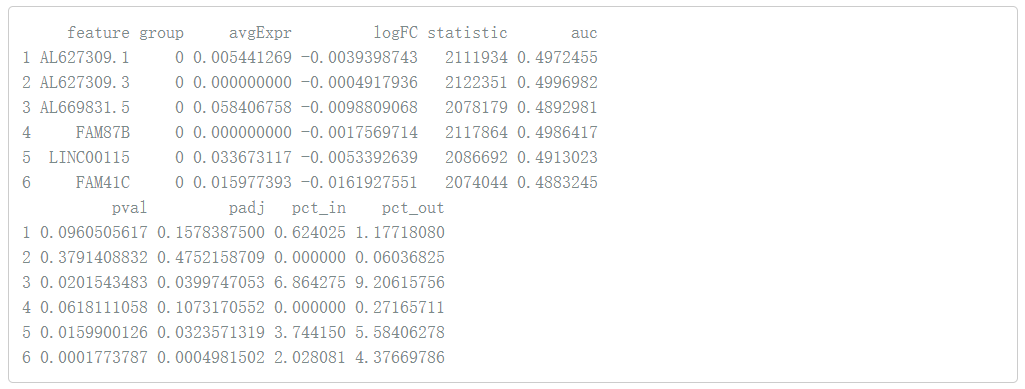
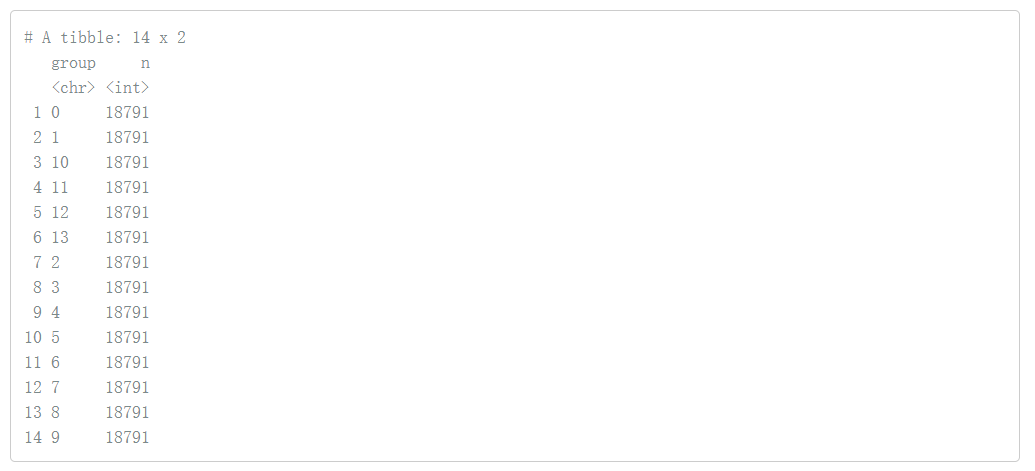
# 我们拥有每个cluster的所有基因 dplyr::count(pbmc.genes, group)
为了进行基因集富集分析,首先需要注释基因集。一个重要的来源是Broad Institute开发的MsigDB[3] ,如下图:
使用fgsea进行基因集富集
library(msigdbr) library(fgsea) library(dplyr) library(ggplot2) msigdbr_show_species()#我们看看都有神马物种的数据

m_df<- msigdbr(species = "Homo sapiens", category = "C7")#我们使用C7免疫基因集 head(m_df)
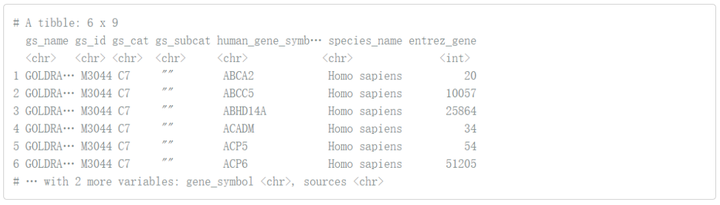
fgsea_sets<- m_df %>% split(x = .$gene_symbol, f = .$gs_name) fgsea_sets$GSE11057_NAIVE_VS_MEMORY_CD4_TCELL_UP [1] "ABLIM1" "ACER1" "ACPL2" "AEBP1" [5] "AGRN" "AIF1" "ALG10B" "AMN1" [9] "ANKRD36BP2" "APBA2" "APBB1" "ARMCX2" [13] "BACH2" "BEND5" "BNIP3L" "BTBD3" [17] "C10orf58" "C12orf23" "C1orf145" "C6orf170" [21] "C6orf48" "CA6" "CADPS2" "CAMK4" [25] "CD248" "CD55" "CENPV" "CEP41" [29] "CHI3L2" "CHML" "CHMP7" "CIAPIN1" [33] "CLCN5" "COL5A2" "CRLF3" "CYHR1" [37] "DDR1" "DFNB59" "DNHD1" "DNTT" [41] "DSC1" "EDAR" "EEA1" "EFNA1" [45] "ENGASE" "EXPH5" "FAM113B" "FAM134B" [49] "FCGRT" "FLJ13197" "GAL3ST4" "GNAI1" [53] "GP5" "GPR125" "GPR160" "GPRASP1" [57] "GPRASP2" "GPRC5B" "HEMGN" "HIPK2" [61] "HSF2" "IGF1R" "IGIP" "ITGA6" [65] "KCNQ5" "KCTD3" "KLHDC1" "KLHL13" [69] "KLHL24" "KRT18" "KRT2" "KRT72" [73] "KRT73" "LINC00282" "LMLN" "LOC100286937" [77] "LOC100287237" "LOC100289019" "LOC100507218" "LOC282997" [81] "LOC283887" "LOC284023" "LOC346887" "LOC439949" [85] "LOC440104" "LOC641518" "LOC644794" "LOC646762" [89] "LOC646808" "LPHN1" "LRRN3" "MALL" [93] "MAML2" "MANSC1" "ME3" "MEF2A" [97] "MEST" "METAP1D" "MIR101-1" "MIR600HG" [101] "MLXIP" "MPP1" "MPP7" "MRPL45P2" [105] "MYB" "MZF1" "NAA16" "NBEA" [109] "NDFIP1" "NET1" "NPAS2" "NPM3" [113] "NSUN5" "NUCB2" "NUDT12" "NUDT17" [117] "PADI4" "PCSK5" "PDE3B" "PDE7A" [121] "PDE7B" "PDE9A" "PDK1" "PECAM1" [125] "PHGDH" "PIGL" "PIK3CD" "PIK3IP1" [129] "PITPNM2" "PKIG" "PLA2G12A" "PLAG1" [133] "PLLP" "PRRT1" "PTPRK" "RAPGEF6" [137] "REG4" "RGS10" "RHPN2" "RIN3" [141] "RNF175" "ROBO3" "SATB1" "SCAI" [145] "SCARB1" "SCML1" "SCML2" "SERPINE2" [149] "SERTAD2" "SERTM1" "SETD1B" "SFMBT2" [153] "SFXN2" "SGK223" "SH3RF3" "SIAH1" [157] "SLC11A2" "SLC25A37" "SLC29A2" "SLC2A11" [161] "SMPD1" "SNORD104" "SNPH" "SNRPN" [165] "SNX9" "SORCS3" "SPPL2B" "SREBF1" [169] "STAP1" "STK17A" "TAF4B" "TARBP1" [173] "TGFBR2" "TIMP2" "TLE2" "TMEM170B" [177] "TMEM220" "TMEM41B" "TMEM48" "TMIGD2" [181] "TOM1L2" "TSPAN3" "TTC28" "TUG1" [185] "UBE2E2" "USP44" "VPS52" "ZC4H2" [189] "ZMYND8" "ZNF182" "ZNF229" "ZNF238" [193] "ZNF496" "ZNF506" "ZNF516" "ZNF546" [197] "ZNF563" "ZNF662" "ZNF780B" "ZNRD1-AS1"
GSE11057_NAIVE_VS_MEMORY_CD4_TCELL_UP代表神马呢?

fgsea()函数需要一个基因集列表以及对应值,主要是基因名和AUC(AUC:ROC曲线下方的面积大小,简单说就是代表准确性,准确性越高,AUC值越大),其中基因名要与pathway中的名字是一样的,不然也看不到富集通路啊。详细参考:https://stephenturner.github.io/deseq-to-fgsea/,这里作者对转录组数据进行了GSEA,作者的部分代码来自与此。
# Naive CD4+ T cells pbmc.genes %>% dplyr::filter(group == "0") %>% arrange(desc(logFC), desc(auc)) %>% head(n = 10) #进行降序排序
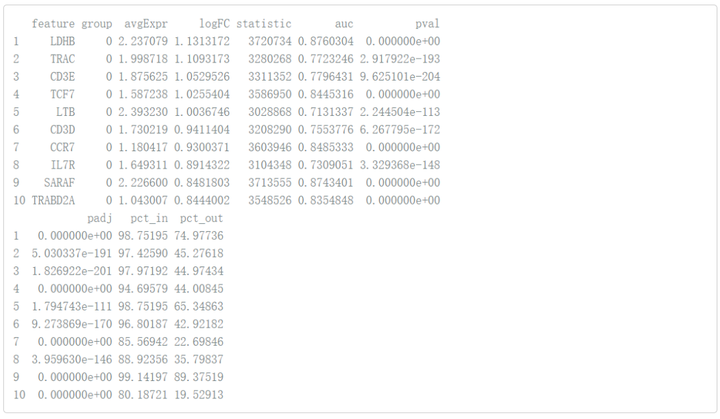
# 在cluster0中我们可以看到IL7R和CCR7,都是幼稚CD4 + T细胞的标记基因 # 仅选择fgsea的feature和auc列 cluster0.genes<- pbmc.genes %>% dplyr::filter(group == "0") %>% arrange(desc(auc)) %>% dplyr::select(feature, auc) ranks<- deframe(cluster0.genes) head(ranks)

看见这些核糖体相关基因我的脑子就是痛痛。。。
fgseaRes<- fgsea(fgsea_sets, stats = ranks, nperm = 1000) Warning in fgsea(fgsea_sets, stats = ranks, nperm = 1000): There are ties in the preranked stats (21% of the list). The order of those tied genes will be arbitrary, which may produce unexpected results. #这也表明你运行了结果可能每次都略有区别;
整理数据:
fgseaResTidy <- fgseaRes %>% as_tibble() %>% arrange(desc(NES)) fgseaResTidy %>% dplyr::select(-leadingEdge, -ES, -nMoreExtreme) %>% arrange(padj) %>% head()
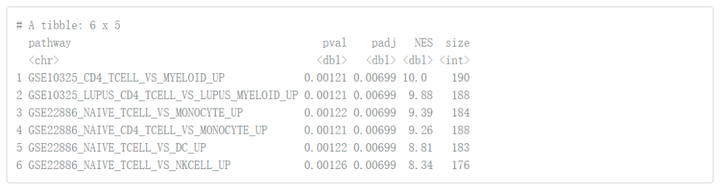
应用标准化富集分数绘制barplot
# 显示top20信号通路 ggplot(fgseaResTidy %>% filter(padj < 0.008) %>% head(n= 20), aes(reorder(pathway, NES), NES)) + geom_col(aes(fill= NES < 7.5)) + coord_flip() + labs(x="Pathway", y="Normalized Enrichment Score", title="Hallmark pathways NES from GSEA") + theme_minimal() ####以7.5进行绘图填色
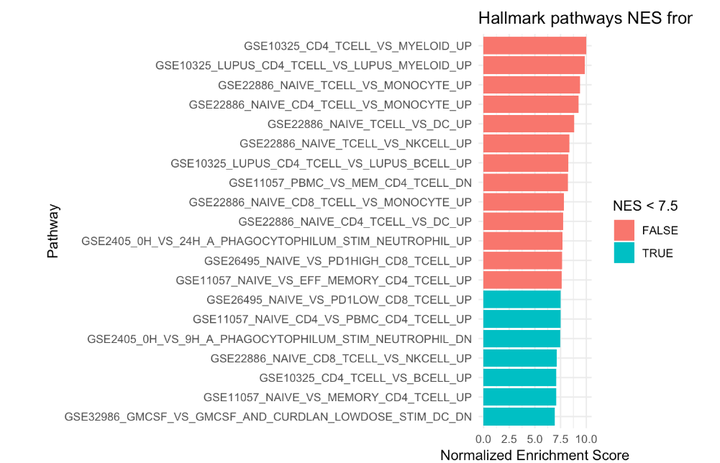
GSEA style plot
plotEnrichment(fgsea_sets[["GSE10325_CD4_TCELL_VS_MYELOID_UP"]], ranks) + labs(title="GSE10325 CD4 TCELL VS MYELOID UP")
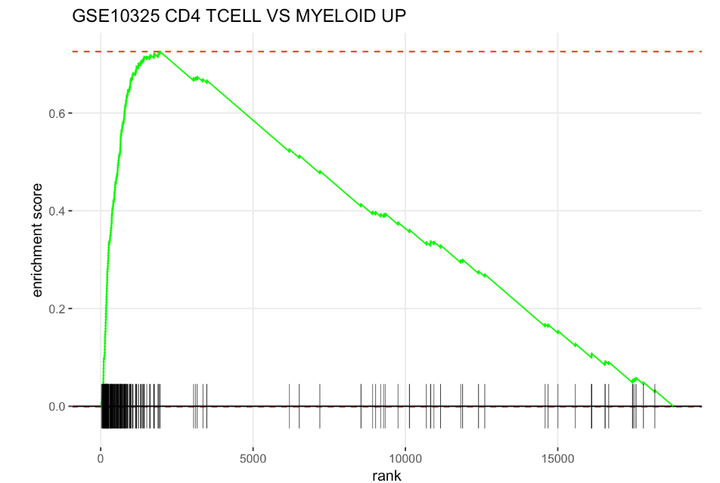
以上图像是个啥意思呢?
X轴是实验中的所有基因(在这种情况下,大约为20,000)。每个黑条是该基因集中的基因(途径)。我们可以知道基因在排序列表中的位置。
如果基因集位于预先排列的基因列表的顶部,则通过某种度量计算出富集分数(enrichment score,ES),ES为正。如果基因集位于预先排列的基因列表的底部,则ES为负。
从以上图中可以看出多数基因都落在了峰值之前(绿线峰值)的核心基因集中,表明基因在该数据集中的显著富集,也就是高表达。
参考
[1]:http://cf.10xgenomics.com/samples/cell-exp/3.0.2/5k_pbmc_v3/5k_pbmc_v3_filtered_feature_bc_matrix.tar.gz
[2]:https://github.com/harvardinformatics/micro-course/blob/3aea680594b7f93b4038b200933c9efc9cda2fa2/scRNAseq/scRNAseq_workshop_1.Rmd[3]:http://software.broadinstitute.org/gsea/msigdb/index.jsp
你可能还想看
https://wap.sciencenet.cn/blog-118204-1241277.html
上一篇:一个R包完成单细胞基因集富集分析 (全代码)
下一篇:图形解读系列 | 给你5个示例,你能看懂常用热图使用吗?