博文
主流衰老理论和干预的可行性论证
||
作者:黄必录
摘要:很多衰老理论都是错误的,因此缺乏有效的干预措施,在这里,我们先简单介绍一下最新的衰老理论,然后对几大主流的衰老理论与干预措施进行可行性论证。
关键词:遗传程序;细胞衰老;rDNA;衰老干预;退行性疾病
本文预印本:https://ssrn.com/abstract=5151210
1 介绍
我们都会慢慢变老,然后死亡,而且很多退行性疾病也是由衰老导致的,因此,与其分别解决不同疾病,不如集中解决衰老问题。虽然人们已经提出了很多衰老理论,但是,很多衰老理论都是错误的,因此缺乏有效的干预措施。为此,本文先简单介绍一下最新的衰老理论,然后对几种主流的衰老理论和干预措施进行可行性论证。
2 衰老的本质是一种程序
由于每种生物都有一个相对固定的生长发育、成熟衰老和死亡的时间表,因此,衰老的本质是一种遗传程序,而不是随机的损伤积累。因为随机的损伤积累无法解释将几种同一属的身体结构极为相似的但寿命不一样的鳉鱼,放在同一水池下人工饲养,它们寿命的差别为什么仍然还存在[1]。
细胞衰老过程中,基因表达也是程序化的,例如,造血干细胞衰老过程会有1500种基因上调,1500种基因下调[2],据此,通常的抗衰老措施就是将某个上调基因进行抑制,某个下调基因进行激活或过表达,但是,这种在代谢层面和信号通路上的干预,只能小幅度延长寿命,而且副作用大,更不可能返老还童。
3 细胞衰老是由端粒DNA和核糖体DNA共同调控的
要让在染色体上固定不变的基因实现程序化表达,必须要有个计时的装置驱动遗传程序的运行,而端粒这种多拷贝的串联重复序列DNA相当于沙漏计时器中的沙子,是“计时物质”的最佳候选者。但是,很多细胞在衰老过程中端粒并没有缩短,甚至还有延长的,一些终末分化细胞的染色体已经不再复制,因此端粒缩短也不明显,据此推测,除了端粒,必然还有另一种多拷贝的串联重复序列DNA作为驱动遗传程序运行的计时物质。为此提出了“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”[3],本理论认为,随着端粒和/或rDNA阵列的逐渐缩短,P53就会沿着时间轴产生“浓度梯度”,由于P53会与多种基因的启动子和增强子结合,从而使有些基因表达上调,有些基因表达下调,以此驱动染色体上的基因群进行程序化表达[1]。
为了验证“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”是否是正确的,我们通过敲低小鼠和人类原代细胞中的 45S rDNA拷贝数,结果:衰老标志物P53、P21、P16和SA-β-GAL都显著上调,端粒长度、细胞活力和细胞传代次数都显著减少。此外还检测了小鼠的衰老细胞和hESC与hiPSC,发现衰老细胞的端粒长度和45S rDNA拷贝数都显著减少了,hESC与hiPSC的端粒长度和45S rDNA拷贝数显著增加了,这些数据有力的证明了hESC和hiPSC的返老还童机制不是因为表观遗传重编程,而是因为端粒和45S rDNA阵列的长度都显著增加了,细胞衰老和Hayflick极限的根本原因是由端粒和 45S rDNA 共同调控的,而且rDNA对衰老的权重大于端粒(未发表的观察)。从第一性原理看,物种寿命是由端粒和/或rDNA阵列的缩短速率决定的。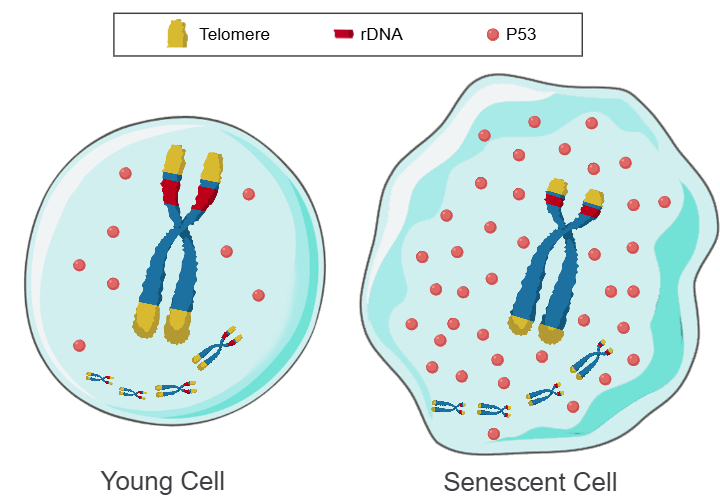
图1:Telomere DNA and ribosomal DNA co-regulation model for cell senescence
左图:染色体上长阵列的端粒和rDNA,P53迅速降解,P53水平低,细胞年轻。
右图:染色体上短阵列的端粒和rDNA,P53缓慢降解,P53水平高,细胞衰老。
一个理论是否正确,要看这个理论是否具备自洽性,既然端粒和rDNA阵列的缩短是导致细胞衰老的根本原因,是驱动基因群程序化表达的计时物质,那么,在体细胞中损耗掉的端粒和rDNA,在生殖细胞或胚胎的早期细胞中必须要能够得到补充,否则,生命无法进行世代轮回。幸运的是,已有证据表明,在体细胞中损耗掉的端粒和rDNA,可以在胚胎早期细胞或生殖细胞中得到补充[4-6]。
小鼠造血干细胞会因为mTOR1激活导致rDNA阵列缩短[7],而抗衰老药雷帕霉素能通过抑制mTOR1来延缓衰老。
4 主流衰老理论和干预措施的可行性论证
一个正确的理论是不容许存在一个漏洞,并且漏洞越多就越不靠谱。
4.1 衰老的表观遗传理论
随着年龄的增长,全基因组DNA甲基化水平会逐渐的下降,据此可以人为赋予一种用来测量生理年龄的表观遗传时钟[8]。2023年,David A. Sinclair等人[9]提出了衰老的信息论(Information Theory of Aging,ITOA),进一步描述了衰老过程中表观遗传信息丢失的类型,包括DNA甲基化减少、转录因子失调、非编码RNA、染色质结构改变、组蛋白修饰和丰度。ITOA理论认为表观遗传信息丢失是驱动衰老的重要原因。
DNA去甲基化和加甲基化、组蛋白去乙酰化和加乙酰化都是同时进行的,因此,表观遗传修饰是动态的不稳定的,不具备计时物质的属性,因此,表观遗传的增龄性变化不可能是驱动衰老的原因,而是由端粒DNA和rDNA的阵列缩短产生的结果[10-12],就象脸上的皱纹不是皮肤衰老的原因,而是皮肤细胞衰老的结果。相比衰老细胞,年轻细胞的端粒更长,端粒酶活性更高,TERT被证明与染色质重塑因子相互作用,并调节DNA甲基化[13]。因此,ITOA是一个错误的理论,并且该理论已遭到另一些人质疑[14-15]。
改变细胞表观遗传的方法称为“表观遗传重编程“,包括“多能重编程”、“部分重编程”和“直接重编程”。在多能重编程中,体细胞失去身份,表观遗传年龄(EpiAge)逆转到0岁。“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”推测[3],重编程使细胞返老还童的根本原因是因为端粒和rDNA阵列的大幅度延长。我们的检测也发现,和衰老细胞相比,hESC和hiPSC的端粒长度和45S rDNA拷贝数都显著增加了,因此,hESC和hiPSC的返老还童机制不是因为表观遗传重编程(未发表的观察)。
由于iPSC和ESC端粒和rDNA阵列不会缩短,因此,这些细胞不会经历表观遗传老化[16],当iPSC和ESC分化为体细胞后,端粒很快开始缩短[17]。由于表观遗传时钟是由端粒和rDNA调控的[10-12],因此,当iPSC和ESC开始分化之时,也是表观遗传时钟很快开始嘀嗒作响之时[18]。但是,已发现72%的hiPSC的DNA受到严重损伤,因此,由iPSC分化的体细胞会被免疫系统清除掉,这将无法用来让衰老的组织返老还童或治疗与衰老有关的退行性疾病;在部分重编程中,这些细胞身份不变,端粒也没延长或略有缩短,一旦停止表达山中因子,衰老症状又会很快积累,包括EpiAge恢复到重编程之前的状态[19];直接重编程又称“转分化”,将老年成纤维细胞直接重编程为神经干细胞,则保留了衰老的表型,EpiAge不会逆转[20],提示,采用直接重编程方法治疗神经退行性疾病可能行不通。
还有很多依据表明衰老的表观遗传理论是错误的,例如:Altos Labs公司,他们通过部分重编程,仅增加野生型小鼠中位寿命12% [21],还不如小分子抗衰老药,增加这点寿命的原因可能与山中因子促进细胞再生有关。例如,促进血管生成能显著延长小鼠寿命[22];多西环素能通过激活ATF4信号通路,在小鼠心肌梗死后促进心脏再生[23],延长秀丽隐杆线虫寿命72.8%,让脂褐素(LF)含量下降了约50%[24],延长早衰小鼠寿命[25];细胞越衰老,DNA甲基化水平越低,而原始生殖细胞(Primordial germ cell, PGC)的基因组也是很少有甲基化的DNA[26],是不是PGC也是最衰老的细胞?其实恰恰相反,PGC和iPS细胞一样具有永垂不朽的潜力;有些动物衰老与甲基化丢失无关,例如,果蝇和双翅目昆虫中的DNA很少或没有发生甲基化。常用来研究衰老的秀丽隐杆线虫也没有甲基化的DNA。Steve Horvath是加州大学洛杉矶分校的人类遗传学和生物统计学教授,他说美国国立卫生研究院一直支持我,但说实话,这还不够。原因是表观遗传时钟有点争议,因为一些研究人员认为,由于蠕虫没有表观遗传时钟,它怎么会衰老?这可能会使拨款申请更加困难;实验显示,在重编程过程中,细胞生理状态的年轻化发生在表观遗传特征的年轻化之前[27],说明重编程导致的衰老逆转与表观遗传改变无关;部分重编程结束后,发现端粒没有延长或略有缩短,衰老标志又开始积累,EpiAge又恢复到重编程之前的状态[19];虽然连续补充α-酮戊二酸(AKG)7个月能使EpiAge拨回8年[28],长期摄入AKG的雌性中年小鼠,中位数寿命虽然延长了近16%,但对雄性小鼠的寿命影响不显著[29];生长激素能逆转EpiAge[30],对老年个体生长激素补充实验发现,生长激素可使61-81岁老人肌肉增加皮肤变厚,年轻10-20岁[31],但实际上长期使用生长激素会加速衰老[32-34],一位60岁的老人接受了生长激素释放激素(GHRH)基因治疗后,EpiAge下调了6岁,但端粒年龄却比同龄人老了7个月[35]。提示,一些能逆转EpiAge,增强线粒体功能和促进细胞增殖的抗衰老药,实际上是在透支有限的细胞分裂次数和寿命,EpiAge只能表示新陈代谢率。
由于端粒DNA和rDNA是很脆弱的串联重复阵列,转录过程很容易使拷贝丢失,而合成蛋白质需要先合成占RNA总量82%的rRNA,因此,生长激素下调EpiAge和缩短寿命的悖论,其实就是促进蛋白质和ATP合成,检测时显示年轻状态,实际上是透支了有限长度的端粒和rDNA的阵列。值得注意的是,很多具有促进蛋白质和ATP合成的所谓的抗衰老药,都有可能透支寿命。例如,增加NAD+的抗衰老药NR能够增加线粒体功能,经最严格的美国国家老龄研究所测试,反而使雄性小鼠寿命缩短3%[36];秀丽隐杆线虫添加抗氧化剂维生素C,线粒体ATP产量增加了2.5倍以上,在治疗后的第8天,LF含量增加了约18%,同时寿命缩短[23]。
随着年龄的增长,全基因组DNA甲基化水平为什么会逐渐的下降?
DNA甲基转移酶DNMT1对于保持DNA甲基化状态起着重要的作用。而P53会结合到dnmt1基因的启动子上[37],因此,随着端粒和rDNA的阵列逐渐缩短,P53水平就会逐渐上调,从而导致了DNMT1基因的表达量逐渐下调,因此降低了全基因组DNA的甲基化水平。
有个先有鸡先有蛋问题,是DNA先甲基化后才导致转录沉默,还是先转录沉默后才导致DNA甲基化?
反义RNA(antisense lncRNA),通常是由编码蛋白质的基因的反义链转录的,并与该基因的mRNA存在序列重叠,占70%的基因均有反义lncRNA,反义lncRNA的转录往往与其基因的正义链转录存在相关性。反义lncRNA的转录时,会使该位点的DNA被DNA主动去甲基化酶TET3识别,从而清除掉该位点的甲基化修饰[38]。因此,是先有转录因子在促进DNA转录,DNA转录时就会去掉甲基,而在缺乏转录因子时,DNA又会被甲基化。
随着年龄的增长,全基因组DNA甲基化水平会虽然会逐渐下降,但是,有些基因如IFNγ、F3、CRAT 和 OGG,在衰老过程中更容易被甲基化,GCR、iNOS 和 TLR2则更容易被去甲基化[39],而这些基因都与炎症相关。阿尔兹海默症患者神经元中淀粉样前蛋白基因启动子区的甲基化程度也会随着年龄的增加而下降,导致该基因表达上升,从而致使神经系统功能紊乱[40]。因此,衰老或神经退行性疾病不是一种“随机的损伤积累”或“随机的表观遗传信息丢失”[9],而是一种有计划的遗传程序。
综上所述,衰老的表观遗传理论是错误的。
4.2 衰老的自由基理论
1956年,Harman提出了衰老的自由基理论[41],该理论至今还被某些人吹棒,然而,到目前为止也没发现由基清除剂能显著延长小鼠的寿命。
自由基主要是由线粒体中产生的活性氧(reactiveoxygenspecies,ROS)),ROS会攻击各种大分子,造成细胞核DNA和mtDNA突变、影响蛋白质在细胞中的流动性[42],导致端粒缩短[43]。例如,人成纤维细胞在40%的氧分压下培养,细胞每次分裂使端粒缩短的速率由原来的90bp增大到500bp,由原来传代45代降到几代[44];中性粒细胞会以ROS依赖性方式诱导成纤维细胞和肝细胞端粒缩短[45]。因此,在理论上,ROS会促进个体衰老。值得注意的是,在因果关系中,ROS只是众多影响衰老速率的因素之一,而不是导致衰老的根本原因。
但是,衰老的自由基理论忽略了生物在亿万年演化过程中,个体和细胞已经具备了一套完善的冗余的防御系统,由ROS造成的各种损伤是能够克服的。首先,在年轻的细胞组织中,被ROS破坏的各种组份能够很快被修复或更新掉,根本构不成危害。另外,个体衰老主要是由成体干细胞复制性衰老导致的,而成体干细胞大部分时间处于静息的状态,并且居住在缺氧的壁龛中,本来产生的ROS不多,例如,在死亡两周的尸体中能提取到活的干细胞[46]。再加上少量的ROS对细胞有着重要的生理作用[47],因此,过度清除掉ROS反而是有害,例如,几种常见的抗氧化剂似乎会增加死亡率[48],再说,细胞中有个平衡ROS的调节功能,当人为提高ROS水平时,就会相应提高清除ROS功能。
下面列出几个不支持衰老的自由基理论的证据:
LF是由溶酶体产生的难以降解的残渣,ROS会促进LF产生,在有丝分裂后的细胞中,LF含量比有丝分裂细胞更高[49],老年人的皮肤会积累大量的LF,产生可见的衰老外观--老年斑。1973年,Tappel发现,用含有抗氧化剂的维生素E的饲料喂养成年老鼠1年,发现神经元LF确定少见了,但死亡率未见减少;维生素C是一种有效的抗氧化剂,虽然维生素C能延长早衰小鼠寿命,但没发现能延长野生型小鼠寿命[50];秀丽隐杆线虫用维生素C处理,LF含量竟增加了约18%,寿命也缩短了[23]。提示,LF不会驱动衰老,抗氧化剂不会延长寿命。
亚甲基蓝(MB)是一种很强的抗氧化剂,能渗透到溶酶体和线粒体等细胞器,但它并没有延长小鼠的平均寿命[51];过表达超氧化物歧化酶(SOD)也不能延长小鼠寿命[52];提高低等动物的ROS反而能延长健康和最大寿命[53];线粒体超氧化物歧化酶sod-2基因的缺失,反而延长了秀丽隐杆线虫的寿命,尽管氧化损伤的蛋白质显著增加[54]。
还有很多证据不支持衰老的自由基理论,本文不一一列出,综上所述,衰老的自由基理论是错误的。
4.3 衰老的线粒体理论
1980年,Miquel等人[55]提出了细胞衰老的线粒体理论, 该理论认为, 由线粒体产生的ROS会导致细胞中的mtDNA、脂类和蛋白质等物质发生氧化损伤, 引起细胞、组织、器官的异常, 最终加速衰老进程。
但是,被ROS破坏的蛋白质能够被降解和更新,突变的mtDNA也会被选择性清除掉[56]。2002年4月1日我在“科技日报”发表的题为《我们能长生不老吗》写到:“异常或失效的线粒体会被溶酶体识别吞食”[57]。溶酶体选择性吞食突变的线粒体称“线粒体自噬”(mitophagy),这一名词由Lemasters于2005年提出[58]。衰老细胞之所以会积累突变的mtDNA,是因为衰老过程中,遗传程序有意的关闭了这种解决方案。
1958年,Yoshida发现[59],在8小时内,密伊乐藻(Elodoa den.se)有细胞核的原生质体中的叶绿体,经历了衰老和结构破坏的过程,而无细胞核原生质体的叶绿体仍保持绿色和连续积累淀粉;1965年,Wright和 Hayflick把年轻的细胞核植入去核的衰老的细胞质中,结果细胞恢复了年轻,并按年轻的细胞核剩余的分裂次数继续分裂下去。相反,把衰老的细胞核植入去核的年轻的细胞质中,结果细胞出现衰老表型,细胞分裂次数大为减少。这些研究表明,决定细胞衰老的部位不是线粒体,而是细胞核。也就是说,mtDNA突变积累不可能会导致细胞衰老。此外,增加端粒长度能显著增加细胞分裂次数,使小鼠中位数寿命延长40%,体毛变黑[60],但没有证据表明,促进线粒功能也能增加细胞分裂次数和显著延长小鼠寿命。
mtDNA突变是导致动物衰老,还是仅仅与之相关,这是一个激烈争论的问题。下面列出几个不支持细胞衰老的线粒体理论的证据:
猕猴和人类的卵母细胞中的mtDNA突变不会随着年龄的增长而积累[61];Vermulst等人[62]发现,增加mtDNA突变的小鼠模型没有任何加速衰老的迹象,也不会缩短寿命;小鼠线粒体超氧化物歧化酶(SOD2)的杂合突变虽然导致氧化损伤和mtDNA突变增加,但并没有缩短动物的寿命[63];线粒体功能障碍的主要特征是ATP的产量下降。然而,用多西环素抑制秀丽隐杆线虫的线粒体产生ATP,治疗组寿命反而延长了72.8%。而抗氧化剂维生素C处理,导致产生的ATP增加了2.5倍,在治疗后的8天,虫体内的LF含量竟增加了约18%,同时加速了衰老[23]。
细胞和个体具备着各种清除突变的mtDNA的解决方案:重编程过程导致mtDNA突变的异质体出现双峰分布,在四次分裂后要么迅速失去了突变,要么获得了更多。具有高突变的异质体的iPSC生长慢,EpiAge上升。提示,在组织中,mtDNA具有高突变的细胞竞争不过健康细胞而会被淘汰掉[64];果蝇可以通过线粒体自噬选择性清除掉肌肉中含有突变的mtDNA的线粒体[65];溶酶体能够降解受损的线粒体,当溶酶体功能受损或不堪重负时,成纤维细胞和心肌细胞会通过外泌体排泄掉损坏的线粒体[66];神经元内损伤的线粒体会选择性排到细胞外,然后被星形胶质细胞摄取降解掉[67];干细胞在分化过程,会把缺陷的线粒体多分配给子细胞的祖细胞,把正常的线粒体多分配给子细胞的干细胞[68]。
还有很多证据不支持细胞衰老的线粒体理论,本文不一一列出,综上所述,衰老的线粒体理论是错误的。
4.4 衰老的体细胞突变理论
1959年,Szilard 提出[69],体细胞的DNA会不断突变和积累,有可能会导致衰老。
但是,该理论忽略了生物在亿万年演化过程中,已经找到了清除个体中突变体细胞的解决方案,首先,个体是由大量的体细胞组成的,这些体细胞不会全部发生突变,而对于DNA突变的细胞:(1)修复;(2)修复不好启动细胞凋亡;(3)如果修复不好也不凋亡,最终会被免疫系统清除掉。因此,DNA突变不是导致个体衰老的原因,之所以会在衰老的个体中积累突变的体细胞,是因为在衰老过程中,遗传程序会有意的关闭掉DNA修复机制和免疫监视机制。再说,有些植物能活了几千年,以及许多能靠无性繁殖的动植物,虽然它们的细胞经历了无数次分裂,但并没有因为DNA突变和积累而灭绝。衰老的体细胞突变理论也无法解释短命的秀丽隐杆线虫和寿命长迖400年的格陵兰鲨,它们的寿命为何有着如此巨大的差别。
下面列出几个不支持个体衰老的体细胞突变积累理论的证据:
衰老细胞的细胞核DNA仅有少量的突变[70];细胞核DNA突变积累并不会加速衰老[71];HeLa细胞的细胞核会迅速积累DNA损伤[72]。然而,HeLa细胞的分裂次数仍然是无限的。
电离辐射会极大的增加DNA突变率,然而,低剂量的电离辐射反而能延长果蝇、家蝇、大鼠和小鼠的寿命 [73-76];原子弹幸存者比平均寿命要长[77]。这些研究都表明,个体衰老的体细胞突变积累理论是错误的。
电离辐射与衰老表型相关,但并不影响端粒长度和EpiAge[78-79],说明辐射造成的衰老表型并非真正的衰老,而是细胞对DNA损伤反应的表现;对小鼠进行慢性、低剂量全身照射导致了肌细胞数量减少、增殖能力降低和静止期增加,但对分化能力几乎没有影响[80],提示,细胞静止期的增加可以延缓到达Hayflick极限的时间,从而延长小鼠寿命;辐射能触发心脏细胞恢复到更健康的状态[81];美国国家航空航天局双胞胎的研究表明,虽然太空飞行会导致一些外周血细胞群体中的端粒延长,但返回地球会导致端粒在48小时内迅速缩短,并在几个月内恢复到接近基线的水平[82]。辐射暴露和肌肉减负相结合也会导致小鼠肌肉干细胞(MuSCs)和肌纤维细胞核中的端粒延长,但没有在停止辐射后观察到端粒缩短,相反,它们保留了已经延长的端粒长度,并继续增加[83]。这些研究可以解释,为什么电离辐射在增加DNA突变率的情况下反而会延长寿命。
不分裂和频繁分裂的细胞,DNA突变率是相同的[84];虽然哺乳动物的不同物种寿命和体型差距很大,但在生命终结时,体细胞突变负荷仅相差约3倍[85];经典认为,DNA突变积累到产生癌症是个缓慢的过程,然而,鳉鱼会在短暂的时间内胸腺迅速变小,有害突变广泛积累和产生癌症[86-87]。这些研究表明,DNA突变的积累速率与细胞分裂次数等因素无关,而可能与免疫系统衰老有关,因为寿命长的物种能够更长时间保持免疫监视的灵敏度,使突变细胞能够及时清除掉。
最近研究认为,DNA损伤积累可能是导致EpiAge增加的原因[88]。我觉得不对,因为HeLa细胞能快速积累突变的DNA[72],但EpiAge不会增加;电离辐射也会导致DNA损伤,但EpiAge不会增加[78-79];果树利用芽变培育新品种就是靠植物的分生组织中的干细胞的基因突变,但植物分生组织中的干细胞EpiAge不会增加[89];iPSC可检测的DNA损伤超过70%[90],但iPSC的EpiAge仍为零岁[79]。
是不是DNA修复能力越强,DNA突变率越低,寿命就越长?不是,因为蟑螂和水熊虫有超强的DNA修复能力,但寿命却比人类短很多。按照“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”推测[3],物种寿命是由端粒和/或rDNA阵列的缩短速率决定的。例如,果蝇在40天寿命里,rDNA阵列会缩短一半[6]。
还有很多证据不支持个体衰老的体细胞突变积累理论,本文不一一列出,综上所述,个体衰老的体细胞突变积累理论是错误的。
4.5 衰老的自噬理论
自噬就是指细胞中各种功能障碍的细胞器和垃圾物质,转运到溶酶体中降解,以实现废物循环利用。有一种衰老理论认为,细胞中积累各种垃圾物质可能会导致衰老和退行性疾病,提高自噬可以干预衰老和退行性疾病。
生物在亿万年演化过程中,已经找到了清除细胞中各种垃圾物质和功能障碍的细胞器的解决方案,衰老的细胞中之所以会积累各种垃圾物质或功能障碍的细胞器,也是因为在衰老过程中,遗传程序有意的关闭了这些解决方案。也就是说,是衰老导致垃圾物质的积累,是衰老导致退行性疾病,而不是垃圾物质积累导致衰老,垃圾物质积累导致神经退行性疾病。例如,阿尔兹海默症(AD)的大脑中会积累大量的LF、错误折叠的β-淀粉样蛋白(Aβ)和过度磷酸化的 tau 蛋白。1973年,Tappel发现,用含有维生素E的饲料喂养成年小鼠1年,发现神经元LF确定少见了,但死亡率未见减少;含蛋白质2%的饲料喂养9-15周,神经系统有大量的LF形成,然后再喂含蛋白质25%的饲料,又减少了LF[91],然而,高蛋白饮食会加速衰老。
抗体能有效清除Aβ,但对AD的症状改善不显著[92-94],说明Aβ等垃圾物质的积累不是导致AD的原因,这就是6000多亿美元研发经费打水漂的原因[95],方向不对,努力白费。
新生的神经元来自于神经干细胞分化的。Aβ主要靠肝脏合成的白蛋白转运到肝脏中降解[96],小胶质细胞也能大量清除Aβ,而小胶质细胞来自于骨髓干细胞分化的,组成血脑屏障中的几种细胞也来自于骨髓干细胞分化的,因此,导致AD的根本原因是大脑中的神经干细胞和远离大脑的肝脏干细胞和骨髓干细胞衰老导致的[97-99],因此,AD无药可救,专注于Aβ和Tau的药物研发注定都会打水漂。
下面列出几个不支持通过增强自噬来干预衰老的证据:
有些情况下增强自噬反而有害健康[100];小鼠卵巢衰老与颗粒细胞自噬增强有关[101];氯喹(CQ,Chloroquine)是是食品药品监督管理局(FDA)批准的唯一的自噬抑制剂[102]。但CQ反而能让大鼠最大寿命延长了13%,这与有史以来在小鼠模型中测试的一些最好的抗衰老药不相上下[103];关闭老年秀丽隐杆线虫自噬,寿命反而延长了50%[104];增加秀丽隐杆线虫肠道自噬反而会加速衰老[105]。
还有很多证据不支持增强自噬能够干预衰老的证据,本文不一一列出,综上所述,通过增强自噬干预衰老的措施是错误的。
4.6 衰老的炎症理论
衰老细胞的细胞核和线粒体都会向细胞质释放dsRNA和cDNA,细胞以为受到病毒感染,就会启动了先天免疫反应,产生衰老相关分泌表型(SASP)和无菌性炎症[106-109],以召唤免疫细胞过来清除掉衰老细胞,因此,在因果关系中,炎症只是细胞衰老的结果,而不是驱动细胞衰老的原因,抑制炎症不利于免疫系统清除衰老细胞,也不会延长个体的寿命甚至还会加速衰老。
下面列出几个不支持通过抑制炎症来干预衰老的证据:
逆转录酶抑制剂(NRTIs)能够抑制细胞核产生cDNA,但没能延长野生型小鼠寿命[110];能提升NAD+的抗衰老药NR能减少52.6%炎症因子的释放[111],巨噬细胞迁移抑制因子拮抗剂(MIF098)可以帮助巨噬细胞转移,降低机体内的慢性炎症,但经最权威的美国国家老龄研究所测试,发现NR反而使雄性小鼠寿命缩短3%,MIF098也不能延长小鼠寿命[36]。
随着年龄的增长,SASP会越来越高,但端粒缩短速率反而越来越慢[112];衰老细胞会释放SASP,清除衰老细胞可以降低SASP,但没发现清除衰老细胞能延长野生型小鼠寿命[113],而且还发现,从青年时期开始清除衰老细胞的雌性小鼠却加速了衰老[114];观察到清除衰老细胞后EpiAge增加,端粒缩短[115];清除衰老细胞会加速老年雌性小鼠卵巢衰老[116]。这是因为清除衰老细胞会刺激周围年轻细胞复制和分化来填充清除掉衰老细胞留下的空穴,从而加速了端粒和rDNA的阵列的缩短,产生复制性衰老。
衰老细胞产生的炎症环境会阻止干细胞增殖,而每日使用衰老细胞清除剂的达沙替尼和槲皮素进行治疗会降低SA-β-gal+细胞数量,减少炎症和加速干细胞增殖[117]。因此,清除衰老细胞,或通过CD36中和减少SASP,会加速干细胞增殖,产生复制性衰老。
与短命的鲨鱼物种相比,参与激活NF-kB信号通路的三个基因家族(TNF、TLR、LRRFIP)的拷贝数在极长寿的格陵兰鲨中显著增加。作者认为,炎症基因显著增加了,导致免疫力增加了,有助于格林兰鲨鱼极长寿[118]。
作为炎症因子IL-11会激活ERK-AMPK-mTORC1信号通路,用抗体抑制 IL-11 虽然能显著延长小鼠寿命[119],但是这不能证明抑制炎症能延长寿命,因为抑制mTORC1本来就能显著延长小鼠寿命,相当于吃mTORC1抑制剂雷帕霉素。
综上所述,衰老细胞和SASP的增加似乎能让干细胞处“静息态”,从而延缓干细胞的复制性衰老。而清除衰老细胞和减少SASP似乎会激活干细胞增殖和分化,促使干细胞的复制性衰老。因此,通过抑制炎症来干预衰老是错误的。
4.7 注射年轻血液抗衰老
吸血鬼疗法是自古以来都有的想法,年轻的血液到底有没有返老还童的功效?
很明显没有,因为实验室天天用小牛血清、胎牛血清培养细胞,细胞照样无法摆脱衰老死亡的命运;给年老小鼠反复注射年轻的血浆,对照组注射生理盐水,结果是,中位数寿命血浆治疗组为26.4月,对照组为27个月[120],年轻血浆组寿命反而略有缩短;注射血液或血浆还会降低NK细胞活力,削弱免疫监视,促进肿瘤细胞的增殖和传播[121];打异体干细胞也会强烈抑制宿主的免疫系统[122]。因此,想用年轻血液、血浆、外泌体和异体干细胞为自己续命,不但不能健康长寿,还会增加患癌概率和缩短寿命。
下面列出一些不支持年轻血液具有抗衰老功效的证据:
衰老的造血干细胞植入年轻的骨髓中,虽然能够将干细胞的整个转录组和基因表达状态逆转回年轻状态,但是,DNA甲基化模式、再生能力和对损伤的修复功能并没有因为年轻的微环境而发生任何改变[123];年轻血液和年轻的微环境不能改善衰老的骨骼干细胞的功能[124]。
通过手术将年轻和年老小鼠血管连接在一起,称“异体共生(heterochronic parabiosis)”,与年轻小鼠异体共生的年老小鼠寿命延长5%,和限制热量可使小鼠的寿命延长27%相比[125],这不值得一提。小幅度的延寿机制可能与年轻小鼠的血浆无关,而是由于年轻小鼠会向年老小鼠提供年轻的骨髓干细胞,这相当于骨髓移植,已发现在异体共生系统中,年老小鼠拥有低于5%的造血干细胞来自于年轻的小鼠[126]。由于骨髓干细胞能够分化为用于补充免疫系统中的各种免疫细胞、血管中的平滑肌细胞和内皮细胞、血脑屏障中的内皮细胞和周细胞、脑内具有清道夫作用的小胶质细胞和脑膜淋巴管的内皮细胞,因此,移植年轻的骨髓能够让免疫系统,循环系统和神经系统等组织保持年轻的状态[98],例如,骨髓移植可替换大脑中90%的小胶质细胞,并显著改善AD症状[99]。值得注意的是,为了避开强烈的免疫排斥,异体共生用的动物都是基因相似度很高的自交系品种,因此无法在人类身上实施,因为在人类中,只有同卵双胞胎拥有相同的基因。
虽然很多研究表明,年轻小鼠的血液能让年老小鼠生理年龄恢复到年轻的状态,让干细胞重新拥有分裂能力,促进肌肉组织和肝细胞的再生[127]。但为什么注射年轻血液的年老小鼠,寿命反而比对照组略有缩短[120]?答案是,年老的小鼠组织会向血液排放SASP和Aβ等等抑制细胞活力的物质,这些有害物质会暂时性的抑制年轻细胞的活力,使生理年龄增加,因此,在停止异体共生后,生理年龄又会重新变小,恢复到基线水平[128]。相反,由于年轻的血液能激发细胞活力,会暂时性的让衰老的细胞产生了返老还童的表型,实际上是透支有限的细胞分裂次数和寿命,这就是注射年轻血浆的老年小鼠寿命反而比对照组略有缩短的原因。因此,延长寿命的方法就是让组织中的成体干细胞处于“静息状”,而非活跃的“增殖态”。
综上所述,通过注射年轻血液和异体干细胞来干预衰老是不可行的。
5 结语
总之,目前主流的衰老理论漏洞百出,从而没有行之有效的干预衰老的措施,从衰老的程序论和第一性原理看,物种寿命是由端粒和/或rDNA阵列的缩短速率决定的,通过 “重建端粒/rDNA阵列” 可以实现细胞年轻化的干预,为抗衰老药物开发指明了方向。
参考文献
[1] Huang B, Hu X. Causality of Aging Hallmarks. Aging Dis. 2025 May 20. doi: 10.14336/AD.2025.0541. Epub ahead of print. PMID: 40423632.
[2] Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010 Apr;45(4):286-90. doi: 10.1016/j.exger.2009.12.010.
[3] Bilu Huang. Telomere DNA and ribosomal DNA co-regulation model for cell senescence. Negative, 2021. 12(3):9-15. doi:10.13276/j.issn.1674-8913.2021.03.003.
[4] Marion RM, Strati K, Li H, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009 Feb 6;4(2):141-54. doi: 10.1016/j.stem.2008.12.010.
[5] Liu L, Bailey SM, Okuka M, et al. Telomere lengthening early in development. Nat Cell Biol. 2007 Dec;9(12):1436-41. doi: 10.1038/ncb1664.
[6] Lu KL, Nelson JO, Watase GJ, et al. Transgenerational dynamics of rDNA copy number in Drosophila male germline stem cells. Elife. 2018 Feb 13;7:e32421. doi: 10.7554/eLife.32421.
[7] Xu B, Li H, Perry JM, et al. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet. 2017 Jun 22;13(6):e1006771. doi: 10.1371/journal.pgen.1006771.
[8] Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. Erratum in: Genome Biol. 2015 May 13;16:96. doi: 10.1186/s13059-015-0649-6.
[9] Lu YR, Tian X, Sinclair DA. The Information Theory of Aging. Nat Aging. 2023 Dec;3(12):1486-1499. doi: 10.1038/s43587-023-00527-6.
[10] Carlund O, Norberg A, Osterman P, et al. DNA methylation variations and epigenetic aging in telomere biology disorders. Sci Rep. 2023 May 16;13(1):7955. doi: 10.1038/s41598-023-34922-1.
[11] Larson K, Yan SJ, Tsurumi A, et al. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet. 2012 Jan;8(1):e1002473. doi: 10.1371/journal.pgen.1002473.
[12] Paredes S, Maggert KA. Ribosomal DNA contributes to global chromatin regulation. Proc Natl Acad Sci U S A. 2009 Oct 20;106(42):17829-34. doi: 10.1073/pnas.0906811106.
[13] Yuan X, Xu D. Telomerase Reverse Transcriptase (TERT) in Action: Cross-Talking with Epigenetics. Int J Mol Sci. 2019 Jul 7;20(13):3338. doi: 10.3390/ijms20133338.
[14] Timmons JA, Brenner C. The information theory of aging has not been tested. Cell. 2024 Feb 29;187(5):1101-1102. doi: 10.1016/j.cell.2024.01.013.
[15] Yang JH, Hayano M, Rajman LA, et al. Response to: The information theory of aging has not been tested. Cell. 2024 Feb 29;187(5):1103-1105. doi: 10.1016/j.cell.2024.01.014.
[16] Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. Erratum in: Genome Biol. 2015 May 13;16:96. doi: 10.1186/s13059-015-0649-6.
[17] Donate LE, Blasco MA. Telomeres in cancer and ageing. Philos Trans R Soc Lond B Biol Sci. 2011 Jan 12;366(1561):76-84. doi: 10.1098/rstb.2010.0291.
[18] Kabacik S, Lowe D, Fransen L, et al. The relationship between epigenetic age and the hallmarks of aging in human cells. Nat Aging. 2022 Jun;2(6):484-493. doi: 10.1038/s43587-022-00220-0.
[19] Gill D, Parry A, Santos F, et al. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. Elife. 2022 Apr 8;11:e71624. doi: 10.7554/eLife.71624.
[20] Mertens J, Paquola ACM, Ku M, et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell. 2015 Dec 3;17(6):705-718. doi: 10.1016/j.stem.2015.09.001.
[21] Sahu SK, Reddy P, Lu J, et al. Targeted partial reprogramming of age-associated cell states improves markers of health in mouse models of aging. Sci Transl Med. 2024 Sep 11;16(764):eadg1777. doi: 10.1126/scitranslmed.adg1777.
[22] Grunewald M, Kumar S, Sharife H, et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science. 2021 Jul 30;373(6554):eabc8479. doi: 10.1126/science.abc8479.
[23] Gao F, Liang T, Lu YW, et al. Reduced Mitochondrial Protein Translation Promotes Cardiomyocyte Proliferation and Heart Regeneration. Circulation. 2023 Dec 5;148(23):1887-1906. doi: 10.1161/CIRCULATIONAHA.122.061192.
[24] Bonuccelli G, Brooks DR, Shepherd S, et al. Antibiotics that target mitochondria extend lifespan in C. elegans. Aging (Albany NY). 2023 Nov 9;15(21):11764-11781.doi:10.18632/aging.205229.
[25] Wang M, Zhang J, Qiu J, et al. Doxycycline decelerates aging in progeria mice. Aging Cell. 2024 Jul;23(7):e14188. doi:10.1111/acel.14188.
[26] Guo F, Yan L, Guo H, et al. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells. Cell. 2015 Jun 4;161(6):1437-52. doi: 10.1016/j.cell.2015.05.015.
[27] Zhang W, Qu J, Liu GH, et al. The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Biol. 2020 Mar;21(3):137-150. doi: 10.1038/s41580-019-0204-5.
[28] Demidenko O, Barardo D, Budovskii V,et al. Rejuvant®, a potential life-extending compound formulation with alpha-ketoglutarate and vitamins, conferred an average 8 year reduction in biological aging, after an average of 7 months of use, in the TruAge DNA methylation test. Aging (Albany NY). 2021 Nov 30;13(22):24485-24499. doi: 10.18632/aging.203736.
[29] Asadi Shahmirzadi A, Edgar D, Liao CY, et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell Metab. 2020 Sep 1;32(3):447-456.e6. doi: 10.1016/j.cmet.2020.08.004.
[30] Fahy GM, Brooke RT, Watson JP, et al. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell. 2019 Dec;18(6):e13028. doi: 10.1111/acel.13028.
[31] Rudman D, Feller AG, Nagraj HS, et al. Effects of human growth hormone in men over 60 years old. N Engl J Med. 1990 Jul 5;323(1):1-6. doi: 10.1056/NEJM199007053230101.
[32] Coschigano KT, Holland AN, Riders ME, et al. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology. 2003 Sep;144(9):3799-810. doi: 10.1210/en.2003-0374.
[33] List EO, Berryman DE, Slyby J, et al. Disruption of Growth Hormone Receptor in Adipocytes Improves Insulin Sensitivity and Lifespan in Mice. Endocrinology. 2022 Oct 1;163(10):bqac129. doi: 10.1210/endocr/bqac129.
[34] List EO, Sackmann-Sala L, Berryman DE, et al. Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR-/-) mouse. Endocr Rev. 2011 Jun;32(3):356-86. doi: 10.1210/er.2010-0009.
[35] Hanley BP, Brewer K, Church G. Results of a 5-Year N-of-1 Growth Hormone Releasing Hormone Gene Therapy Experiment. Rejuvenation Res. 2021 Dec;24(6):424-433. doi: 10.1089/rej.2021.0036.
[36] Harrison DE, Strong R, Reifsnyder P, et al. 17-a-estradiol late in life extends lifespan in aging UM-HET3 male mice; nicotinamide riboside and three other drugs do not affect lifespan in either sex. Aging Cell. 2021 May;20(5):e13328. doi: 10.1111/acel.13328. Epub 2021 Mar 31. Erratum in: Aging Cell. 2022 Nov;21(11):e13672. doi: 10.1111/acel.13672.
[37] Peterson EJ, Bögler O, Taylor SM. p53-mediated repression of DNA methyltransferase 1 expression by specific DNA binding. Cancer Res. 2003 Oct 15;63(20):6579-82.
[38] Canzio D, Nwakeze CL, Horta A, et al. Antisense lncRNA Transcription Mediates DNA Demethylation to Drive Stochastic Protocadherin α Promoter Choice. Cell. 2019 Apr 18;177(3):639-653.e15. doi: 10.1016/j.cell.2019.03.008.
[39] Madrigano J, Baccarelli A, Mittleman MA, et al. Aging and epigenetics: longitudinal changes in gene-specific DNA methylation. Epigenetics. 2012 Jan 1;7(1):63-70. doi: 10.4161/epi.7.1.18749.
[40] Tohgi H, Utsugisawa K, Nagane Y, et al. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999 Jul 5;70(2):288-92. doi: 10.1016/s0169-328x(99)00163-1.
[41] HARMAN D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956 Jul;11(3):298-300. doi: 10.1093/geronj/11.3.298.
[42] Dall'Agnese A, Zheng MM, Moreno S, et al. Proteolethargy is a pathogenic mechanism in chronic disease. Cell. 2024 Nov 25:S0092-8674(24)01274-1. doi: 10.1016/j.cell.2024.10.051.
[43] von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002 Jul;27(7):339-44. doi: 10.1016/s0968-0004(02)02110-2.
[44] von Zglinicki T, Saretzki G, Döcke W, et al. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp Cell Res. 1995 Sep;220(1):186-93. doi: 10.1006/excr.1995.1305.
[45] Lagnado A, Leslie J, Ruchaud-Sparagano MH, et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 2021 May 3;40(9):e106048. doi: 10.15252/embj.2020106048.
[46] Latil M, Rocheteau P, Châtre L, et al. Skeletal muscle stem cells adopt a dormant cell state post mortem and retain regenerative capacity. Nat Commun. 2012 Jun 12;3:903. doi: 10.1038/ncomms1890.
[47] Finkel T. Reactive oxygen species and signal transduction. IUBMB Life. 2001 Jul;52(1-2):3-6. doi: 10.1080/15216540252774694.
[48] Bjelakovic G, Nikolova D, Gluud LL, et al. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2012 Mar 14;2012(3):CD007176. doi: 10.1002/14651858.CD007176.pub2.
[49] Sitte N, Merker K, Grune T, et al. Lipofuscin accumulation in proliferating fibroblasts in vitro: an indicator of oxidative stress. Exp Gerontol. 2001 Mar;36(3):475-86. doi: 10.1016/s0531-5565(00)00253-9.
[50] Massip L, Garand C, Paquet ER, et al. Vitamin C restores healthy aging in a mouse model for Werner syndrome. FASEB J. 2010 Jan;24(1):158-72. doi: 10.1096/fj.09-137133.
[51] Harrison DE, Strong R, Allison DB, et al. Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell. 2014 Apr;13(2):273-82. doi: 10.1111/acel.12170.
[52] Huang TT, Carlson EJ, Gillespie AM, Shi Y, Epstein CJ. Ubiquitous overexpression of CuZn superoxide dismutase does not extend life span in mice. J Gerontol A Biol Sci Med Sci. 2000 Jan;55(1):B5-9. doi: 10.1093/gerona/55.1.b5.
[53] Sanz A. Mitochondrial reactive oxygen species: Do they extend or shorten animal lifespan? Biochim Biophys Acta. 2016 Aug;1857(8):1116-1126. doi: 10.1016/j.bbabio.2016.03.018.
[54] Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009 Feb;5(2):e1000361. doi: 10.1371/journal.pgen.1000361.
[55] Miquel J, Economos AC, Fleming J, et al. Mitochondrial role in cell aging. Exp Gerontol. 1980;15(6):575-91. doi: 10.1016/0531-5565(80)90010-8.
[56] Bilu Huang. The mechanism significance and treatment of aging[M]. Yanjing Medical Correspondence.1998(7.8):1050-1064.
https://m.doc88.com/p-94754096894296.html#
[57] Bilu Huang..我们能长生不老吗[N].科技日报,2002-4-1.
[58] Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005 Spring;8(1):3-5. doi: 10.1089/rej.2005.8.3.
[59] Yoshida Y. On some characteristics of the idioblast in Elodea leaf. J Fac Sci, Niigata Univ Ser. II 1958; 2: 173–178.。
[60] Jaijyan DK, Selariu A, Cruz-Cosme R, et al. New intranasal and injectable gene therapy for healthy life extension. Proc Natl Acad Sci U S A. 2022 May 17;119(20):e2121499119. doi: 10.1073/pnas.2121499119. Epub 2022 May 10. Erratum in: Proc Natl Acad Sci U S A. 2022 Aug 30;119(35):e2212836119. doi: 10.1073/pnas.2212836119. Erratum in: Proc Natl Acad Sci U S A. 2023 Aug 8;120(32):e2311483120. doi: 10.1073/pnas.2311483120.
[61] Arbeithuber B, Anthony K, Higgins B, et al. Mitochondrial DNA mutations in human oocytes undergo frequency-dependent selection but do not increase with age. bioRxiv [Preprint]. 2024 Dec 12:2024.12.09.627454. doi: 10.1101/2024.12.09.627454.
[62] Vermulst M, Bielas JH, Kujoth GC, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007 Apr;39(4):540-3. doi: 10.1038/ng1988.
[63] Van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003 Dec 16;16(1):29-37. doi: 10.1152/physiolgenomics.00122.2003.
[64] Vandiver AR, Torres A Jr, Sanden A, et al. Increased mitochondrial mutation heteroplasmy induces aging phenotypes in pluripotent stem cells and their differentiated progeny. Aging Cell. 2024 Dec 16:e14402. doi: 10.1111/acel.14402.
[65] Kandul NP, Zhang T, Hay BA, Guo M. Selective removal of deletion-bearing mitochondrial DNA in heteroplasmic Drosophila. Nat Commun. 2016 Nov 14;7:13100. doi: 10.1038/ncomms13100.
[66] Liang W, Sagar S, Ravindran R, et al. Mitochondria are secreted in extracellular vesicles when lysosomal function is impaired. Nat Commun. 2023 Aug 18;14(1):5031. doi: 10.1038/s41467-023-40680-5.
[67] Davis CH, Kim KY, Bushong EA, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014 Jul 1;111(26):9633-8. doi: 10.1073/pnas.1404651111.
[68] Katajisto P, Döhla J, Chaffer CL, et al. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015 Apr 17;348(6232):340-3. doi: 10.1126/science.1260384.
[69] Szilard L. ON THE NATURE OF THE AGING PROCESS. Proc Natl Acad Sci U S A. 1959 Jan;45(1):30-45. doi: 10.1073/pnas.45.1.30.
[70] De Majo F, Martens L, Hegenbarth JC, et al. Genomic instability in the naturally and prematurely aged myocardium. Proc Natl Acad Sci U S A. 2021 Sep 7;118(36):e2022974118. doi: 10.1073/pnas.2022974118.
[71] Robinson PS, Coorens THH, Palles C, et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat Genet. 2021 Oct;53(10):1434-1442. doi: 10.1038/s41588-021-00930-y.
[72] Liu Y, Mi Y, Mueller T, et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat Biotechnol. 2019 Mar;37(3):314-322. doi: 10.1038/s41587-019-0037-y.
[73] Lamb, MJ. The effects of radiation on the longevity of female Drosophila subobscura. Journal of Insect Physiology, 1964. 10(3): p. 487-497. doi.org/10.1016/0022-1910(64)90072-1.
[74] Allen RG, Sohal RS. Life-lengthening effects of gamma-radiation on the adult housefly, Musca domestica. Mech Ageing Dev. 1982 Dec;20(4):369-75. doi: 10.1016/0047-6374(82)90104-x.
[75] Caratero A, Courtade M, Bonnet L, et al. Effect of a continuous gamma irradiation at a very low dose on the life span of mice. Gerontology. 1998;44(5):272-6. doi: 10.1159/000022024.
[76] Calabrese EJ, Baldwin LA. The effects of gamma rays on longevity. Biogerontology. 2000;1(4):309-19. doi: 10.1023/a:1026510001286.
[77] Sutou S. Low-dose radiation from A-bombs elongated lifespan and reduced cancer mortality relative to un-irradiated individuals. Genes Environ. 2018 Dec 19;40:26. doi: 10.1186/s41021-018-0114-3. Erratum in: Genes Environ. 2019 Apr 19;41:12. doi: 10.1186/s41021-019-0127-6.
[78] Kabacik S, Lowe D, Fransen L, et al. The relationship between epigenetic age and the hallmarks of aging in human cells. Nat Aging. 2022 Jun;2(6):484-493. doi: 10.1038/s43587-022-00220-0.
[79]Koch CM, Reck K, Shao K, et al. Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res. 2013 Feb;23(2):248-59. doi: 10.1101/gr.141945.112.
[80] Masuda S, Hisamatsu T, Seko D, et al. Time- and dose-dependent effects of total-body ionizing radiation on muscle stem cells. Physiol Rep. 2015 Apr;3(4):e12377. doi: 10.14814/phy2.12377.
[81] Zhang DM, Navara R, Yin T, et al. Cardiac radiotherapy induces electrical conduction reprogramming in the absence of transmural fibrosis. Nat Commun. 2021 Sep 24;12(1):5558. doi: 10.1038/s41467-021-25730-0.
[82] Garrett-Bakelman FE, Darshi M, Green SJ, et al. The NASA Twins Study: A multidimensional analysis of a year-long human spaceflight. Science. 2019 Apr 12;364(6436):eaau8650. doi: 10.1126/science.aau8650.
[83] Tichy ED, Lee JH, Li G, et al. Impacts of radiation exposure, hindlimb unloading, and recovery on murine skeletal muscle cell telomere length. NPJ Microgravity. 2023 Sep 15;9(1):76. doi: 10.1038/s41526-023-00303-1.
[84] Abascal F, Harvey LMR, Mitchell E, et al. Somatic mutation landscapes at single-molecule resolution. Nature. 2021 May;593(7859):405-410. doi: 10.1038/s41586-021-03477-4.
[85] Cagan A, Baez-Ortega A, Brzozowska N, et al. Somatic mutation rates scale with lifespan across mammals. Nature. 2022 Apr;604(7906):517-524. doi: 10.1038/s41586-022-04618-z.
[86] Cui R, Medeiros T, Willemsen D, et al. Relaxed Selection Limits Lifespan by Increasing Mutation Load. Cell. 2019 Jul 11;178(2):385-399.e20. doi: 10.1016/j.cell.2019.06.004. Epub 2019 Jun 27. Erratum in: Cell. 2020 Mar 19;180(6):1272-1279. doi: 10.1016/j.cell.2020.02.038.
[87] Willemsen D, Cui R, Reichard M, et al. Intra-species differences in population size shape life history and genome evolution. Elife. 2020 Sep 1;9:e55794. doi: 10.7554/eLife.55794. PMID: 32869739; PMCID: PMC7462614.
[88] Spencer Chapman M, Mitchell E, Yoshida K, et al. Prolonged persistence of mutagenic DNA lesions in somatic cells. Nature. 2025 Jan 15. doi: 10.1038/s41586-024-08423-8.
[89] Dai D, Chen K, Tao J, et al. Aging drives a program of DNA methylation decay in plant organs. bioRxiv [Preprint]. 2024 Nov 5:2024.11.04.621941. doi: 10.1101/2024.11.04.621941.
[90] Rouhani FJ, Zou X, Danecek P,et al. Substantial somatic genomic variation and selection for BCOR mutations in human induced pluripotent stem cells. Nat Genet. 2022 Sep;54(9):1406-1416. doi: 10.1038/s41588-022-01147-3.
[91] Manocha SL, Sharma SP. Reversibility of lipofuscin accumulation caused by protein malnutrition in the motor cortex of squirrel monkeys, Saimiri scireus. Acta Histochem. 1977;58(2):219-31. doi: 10.1016/S0065-1281(77)80132-3.
[92] van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in Early Alzheimer's Disease. N Engl J Med. 2023 Jan 5;388(1):9-21. doi: 10.1056/NEJMoa2212948.
[93] Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer's Disease. J Prev Alzheimers Dis. 2022;9(2):197-210. doi: 10.14283/jpad.2022.30.
[94] Sims JR, Zimmer JA, Evans CD, et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 2023 Aug 8;330(6):512-527. doi: 10.1001/jama.2023.13239.
[95] New Report Expresses Optimism Over Current State of Alzheimer's Drug Development. Retrieved December 4, 2018, from
https://www.biospace.com/article/pharma-industry-report-on-alzheimer-s-research-in-the-u-s-cautiously-optimistic/
[96] Cheng Y, He CY, Tian DY, et al. Physiological β-amyloid clearance by the liver and its therapeutic potential for Alzheimer's disease. Acta Neuropathol. 2023 Jun;145(6):717-731. doi: 10.1007/s00401-023-02559-z.
[97] Lin S, Nascimento EM, Gajera CR, et al. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature. 2018 Apr;556(7700):244-248. doi: 10.1038/s41586-018-0004-7.
[98] 黄必录.探讨骨髓干细胞衰老与阿尔茨海默病等衰老性疾病的关系[J].医学理论与实践, 2022 ,35 (6 ):933-935. doi: 10.19381/j.issn.1001-7585.2022.06.010.
[99] Yoo Y, Neumayer G, Shibuya Y, et al. A cell therapy approach to restore microglial Trem2 function in a mouse model of Alzheimer's disease. Cell Stem Cell. 2023 Aug 3;30(8):1043-1053.e6. doi: 10.1016/j.stem.2023.07.006. Erratum in: Cell Stem Cell. 2023 Oct 5;30(10):1392. doi: 10.1016/j.stem.2023.08.011.
[100] Zhou B, Kreuzer J, Kumsta C, et al. Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension. Cell. 2019 Apr 4;177(2):299-314.e16. doi: 10.1016/j.cell.2019.02.013.
[101] Li F, Zhu J, Liu J, et al. Targeting Estrogen Receptor Beta Ameliorates Diminished Ovarian Reserve via Suppression of the FOXO3a/Autophagy Pathway. Aging Dis. 2024 Feb 25. doi: 10.14336/AD.2024.0221.
[102] Manic G, Obrist F, Kroemer G, et al. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol. 2014 Jul 15;1(1):e29911. doi: 10.4161/mco.29911.
[103] Li W, Zou Z, Cai Y, et al. Low-dose chloroquine treatment extends the lifespan of aged rats. Protein Cell. 2022 Jun;13(6):454-461. doi: 10.1007/s13238-021-00903-1. Erratum in: Protein Cell. 2024 Apr 1;15(4):313. doi: 10.1093/procel/pwad053.
[104] Wilhelm T, Byrne J, Medina R,et al . Neuronal inhibition of the autophagy nucleation complex extends life span in post-reproductive C. elegans. Genes Dev. 2017 Aug 1;31(15):1561-1572. doi: 10.1101/gad.301648.117.
[105] Ezcurra M, Benedetto A, Sornda T, et al. C. elegans Eats Its Own Intestine to Make Yolk Leading to Multiple Senescent Pathologies. Curr Biol. 2018 Aug 20;28(16):2544-2556.e5. doi: 10.1016/j.cub.2018.06.035. Epub 2018 Aug 9. Erratum in: Curr Biol. 2018 Oct 22;28(20):3352. doi: 10.1016/j.cub.2018.10.003.
[106]Zecchini V, Paupe V, Herranz-Montoya I, et al. Fumarate induces vesicular release of mtDNA to drive innate immunity. Nature. 2023 Mar;615(7952):499-506. doi: 10.1038/s41586-023-05770-w.
[107]López-Polo V, Maus M, Zacharioudakis E, et al. Release of mitochondrial dsRNA into the cytosol is a key driver of the inflammatory phenotype of senescent cells. Nat Commun. 2024 Aug 27;15(1):7378. doi: 10.1038/s41467-024-51363-0.
[108]Zhou X, Singh M, Sanz Santos G, et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021 Dec 1;11(12):3090-3105. doi: 10.1158/2159-8290.CD-20-1741.
[109]Mao J, Zhang Q, Zhuang Y, et al. Reactivation of senescence-associated endogenous retroviruses by ATF3 drives interferon signaling in aging. Nat Aging. 2024 Dec;4(12):1794-1812. doi: 10.1038/s43587-024-00745-6.
[110]Partridge L, Fuentealba M, Kennedy BK. The quest to slow ageing through drug discovery. Nat Rev Drug Discov. 2020 Aug;19(8):513-532. doi: 10.1038/s41573-020-0067-7.
[111]Norheim KL, Ben Ezra M, Heckenbach I, et al. Effect of nicotinamide riboside on airway inflammation in COPD: a randomized, placebo-controlled trial. Nat Aging. 2024 Dec;4(12):1772-1781. doi: 10.1038/s43587-024-00758-1.
[112]Iwama H, Ohyashiki K, Ohyashiki JH, et al. Telomeric length and telomerase activity vary with age in peripheral blood cells obtained from normal individuals. Hum Genet. 1998 Apr;102(4):397-402. doi: 10.1007/s004390050711.
[113]Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011 Nov 2;479(7372):232-6. doi: 10.1038/nature10600.
[114]Fang Y, Medina D, Stockwell R, et al. Sexual dimorphic metabolic and cognitive responses of C57BL/6 mice to Fisetin or Dasatinib and quercetin cocktail oral treatment. Geroscience. 2023 Oct;45(5):2835-2850. doi: 10.1007/s11357-023-00843-0.
[115] Lee E, Carreras-Gallo N, Lopez L, et al. Exploring the effects of Dasatinib, Quercetin, and Fisetin on DNA methylation clocks: a longitudinal study on senolytic interventions. Aging (Albany NY). 2024 Feb 22;16(4):3088-3106. doi: 10.18632/aging.205581.
[116] Xia X, Yang Y, Liu P, et al. The senolytic drug ABT-263 accelerates ovarian aging in older female mice. Sci Rep. 2024 Oct 5;14(1):23178. doi: 10.1038/s41598-024-73828-4.
[117] Moiseeva V, Cisneros A, Sica V, Deryagin O, Lai Y, et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature. 2023 Jan;613(7942):169-178. doi: 10.1038/s41586-022-05535-x. Epub 2022 Dec 21. Erratum in: Nature. 2023 Feb;614(7949):E45. doi: 10.1038/s41586-023-05765-7.
[118] Kaiqiao Yang, Kazuya Nishiwaki, Hideaki Mizobata et al, The Greenland shark genome: insights into deep-sea ecology and lifespan extremes. bioRxiv 2025.02.19.638963; doi: https://doi.org/10.1101/2025.02.19.638963
[119] Widjaja AA, Lim WW, Viswanathan S, et al. Inhibition of IL-11 signalling extends mammalian healthspan and lifespan. Nature. 2024 Aug;632(8023):157-165. doi: 10.1038/s41586-024-07701-9.
[120] Shytikov D, Balva O, Debonneuil E, et al. Aged mice repeatedly injected with plasma from young mice: a survival study. Biores Open Access. 2014 Oct 1;3(5):226-32. doi: 10.1089/biores.2014.0043.
[121] Goubran HA, Elemary M, Radosevich M, et al. Impact of Transfusion on Cancer Growth and Outcome. Cancer Growth Metastasis. 2016 Mar 13;9:1-8. doi: 10.4137/CGM.S32797.
[122] Kurtzberg J, Prockop S, Teira P, et al. Allogeneic human mesenchymal stem cell therapy (remestemcel-L, Prochymal) as a rescue agent for severe refractory acute graft-versus-host disease in pediatric patients. Biol Blood Marrow Transplant. 2014 Feb;20(2):229-35. doi: 10.1016/j.bbmt.2013.11.001.
[123] Kuribayashi W, Oshima M, Itokawa N, et al. Limited rejuvenation of aged hematopoietic stem cells in young bone marrow niche. J Exp Med. 2021 Mar 1;218(3):e20192283. doi: 10.1084/jem.20192283.
[124] Ambrosi TH, Marecic O, McArdle A, et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature. 2021 Sep;597(7875):256-262. doi: 10.1038/s41586-021-03795-7.
[125] Zhang B, Lee DE, Trapp A, et al. Multi-omic rejuvenation and life span extension on exposure to youthful circulation. Nat Aging. 2023 Aug;3(8):948-964. doi: 10.1038/s43587-023-00451-9.
[126] Ma S, Wang S, Ye Y, et al. Heterochronic parabiosis induces stem cell revitalization and systemic rejuvenation across aged tissues. Cell Stem Cell. 2022 Jun 2;29(6):990-1005.e10. doi: 10.1016/j.stem.2022.04.017.
[127] Conboy IM, Conboy MJ, Wagers AJ, et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005 Feb 17;433(7027):760-4. doi: 10.1038/nature03260.
[128] Poganik JR, Zhang B, Baht GS, et al. Biological age is increased by stress and restored upon recovery. Cell Metab. 2023 May 2;35(5):807-820.e5. doi: 10.1016/j.cmet.2023.03.015.
https://wap.sciencenet.cn/blog-3440171-1491253.html
上一篇:回复国内外专家对细胞衰老的共调控模型的质疑
下一篇:衰老标志的因果关系