
红细胞作为主要葡萄糖汇,改善高海拔环境下的葡萄糖耐量
研究亮点
1. 高海拔居民与缺氧小鼠的葡萄糖清除能力均得到提升
2. 缺氧状态下红细胞数量增加,是缺氧性低血糖发生的必要且充分条件
3. 缺氧红细胞中糖酵解代谢体的解离,促进2,3-二磷酸甘油酸的合成增加
4. 吸入性缺氧干预与小分子缺氧模拟物,可改善1型和2型糖尿病模型小鼠的高血糖症状
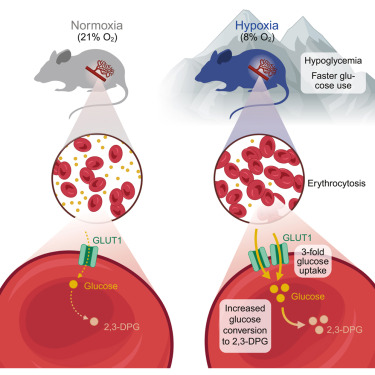
研究摘要
高海拔环境能改善机体葡萄糖耐量、降低糖尿病发病风险,但其潜在的生理机制尚未明确。本研究以小鼠为模型发现,单纯缺氧即可显著提升葡萄糖耐量,且该效应在小鼠恢复正常氧环境后仍能持续数周。正电子发射断层扫描/计算机断层扫描(PET/CT)成像结果显示,除主要内脏器官外,存在一个未知的重要葡萄糖汇。据此我们提出假说:缺氧诱导产生的红细胞是该葡萄糖汇的核心载体。通过放血或输血调控小鼠红细胞数量后,血糖水平发生直接改变,证实红细胞是介导缺氧改善葡萄糖耐量的必要且充分条件。慢性缺氧状态下,红细胞的葡萄糖摄取量持续增加约3倍,葡萄糖转运蛋白1(GLUT1)的蛋白表达量升高约2倍,且该变化特异性发生在新生红细胞中,最终推动糖酵解通量增加,促进2,3-二磷酸甘油酸(2,3-DPG)合成。从作用机制来看,急性缺氧时,脱氧血红蛋白通过竞争性结合,使甘油醛-3-磷酸脱氢酶(GAPDH)从与带3蛋白的抑制性结合中解离,进而增强糖酵解通量,驱动2,3-DPG合成。研究还发现,缺氧或本研究研发的小分子缺氧模拟物HypoxyStat,可逆转1型和2型糖尿病模型小鼠的高血糖症状。本研究证实红细胞是全身葡萄糖代谢的关键调控因子,为高血糖相关疾病提出了全新的治疗思路。
研究引言
大量流行病学研究表明,高海拔环境下人群的糖尿病发病率降低,血糖控制能力提升(表1),这一现象并非人类独有。例如,适应高海拔环境的鹿鼠,其葡萄糖清除能力显著优于低海拔种群;西藏藏猪的胰岛素敏感性高于低海拔猪种,血浆葡萄糖浓度也更低;多种高海拔鸣禽同样表现出血糖水平降低、胰岛素敏感性提升的特征。这些跨物种的研究结果表明,生物体内存在一种进化上保守的生理机制,可在高海拔环境下优化葡萄糖清除过程。
表1 高海拔与血糖控制改善相关的人体研究汇总:已发表的关于慢性高海拔暴露与人体血糖控制相关性的研究,记录了血糖水平、葡萄糖耐量、糖尿病发病风险或高血糖风险等指标(如有相关数据),并标注了各研究的具体海拔、样本量和地理位置。OR=比值比。
高海拔环境下多种生理指标会同时发生变化,难以明确具体是何种因素主导了葡萄糖稳态的改善。针对高海拔人群的研究往往缺乏合适的对照,导致因果关系难以界定,但多项证据表明,氧供不足(缺氧)是核心关键因素。对该现象最早的系统性研究,可追溯至20世纪20至40年代哈佛大学疲劳实验室的开创性工作,该实验室旨在研究极端环境下的人体生理变化,为战时士兵的健康保障提供依据。研究人员在智利安第斯山脉(海拔最高达6000米)开展的一系列精密实验中发现,健康志愿者的葡萄糖耐量在高海拔环境下得到显著改善。这一奠基性发现,在近期的研究中得到了进一步验证和拓展。例如,本团队近期研究证实,慢性低氧暴露的小鼠血糖浓度显著降低,且未出现明显的生理或行为异常。
尽管高海拔环境改善葡萄糖耐量的现象已被广泛证实,但其背后具体的生理机制仍不明确。目前主流假说认为,缺氧条件下外周组织的葡萄糖消耗增加,是该有益效应的成因。事实上,急性缺氧可通过缺氧诱导因子(HIF)的转录应答,快速上调外周组织的葡萄糖转运蛋白表达,进而增强葡萄糖摄取。但随着缺氧暴露时间延长,这一急性信号应答会因负反馈调节而减弱,因此难以单独用该机制解释慢性缺氧下血糖控制的持续性改善。此外,血糖水平在数周内逐步改善的特征,也表明更缓慢的慢性适应性变化,可能是该代谢获益的重要原因。因此,高海拔环境下长期血糖改善的机制,仍是该领域悬而未决的难题。
红细胞是人体中数量最多的细胞类型,约占人体总细胞数的85%,占体重的4%左右。慢性缺氧状态下,红细胞数量可近乎翻倍,占总血容量的比例高达70%以上。成熟红细胞是高度特化的细胞,无细胞核、线粒体等细胞器,主要由血红蛋白构成,核心功能是维持组织的氧供平衡。由于缺乏线粒体,红细胞的能量代谢高度依赖葡萄糖的无氧酵解来合成三磷酸腺苷(ATP)。有趣的是,缺氧状态下血红蛋白的关键变构调节因子——2,3-二磷酸甘油酸(2,3-DPG),作为糖酵解中间产物,可通过卢布林-拉波波特(LR)支路在红细胞内持续合成。缺氧时2,3-DPG水平升高,能促进血红蛋白向组织释放氧气,这一代谢转换在高海拔缺氧暴露后数分钟内即可发生,且能持续数周。这些发现表明,红细胞的葡萄糖代谢与缺氧生理适应之间存在密切联系,也引出一个有趣的推测:低氧环境下,红细胞的葡萄糖利用可能在全身葡萄糖稳态调控中发挥核心作用。
据此,本研究提出假说:缺氧条件下,红细胞是机体的主要葡萄糖汇。为验证该假说,我们利用多种经实验调控红细胞数量的小鼠模型,系统分析了红细胞丰度与血糖水平的关联。研究结果表明,缺氧状态下红细胞丰度的增加及其代谢模式的改变,可解释大部分观测到的血糖变化。本研究揭示了红细胞在全身葡萄糖稳态调控中意想不到的核心作用,为糖尿病治疗开辟了新方向。
研究结果
高海拔居民的葡萄糖耐量改善特征,在缺氧小鼠中成功重现
高海拔环境的特征是多种环境因素共同改变,包括低温、紫外线辐射增强、湿度降低以及氧供不足。为明确高海拔居民的葡萄糖耐量改善是否由缺氧单独介导,我们采用常压低氧暴露的小鼠模型展开研究。将8周龄雄性小鼠分别置于常氧(21% O₂)或低氧(8% O₂,对应海拔5000米以上)环境中饲养3周,定期监测其血糖和体重。小鼠在8% O₂暴露的最初数小时/数天内会出现急性应激反应(如短暂活动减少、摄食抑制、体重下降),但该应激会在适应1周后缓解。研究发现,从低氧暴露第2天开始,小鼠的基础血糖水平显著降低,同时伴随急性体重下降,且体重在1周内趋于稳定。这种早期的血糖降低并非由摄食减少导致,因为与低氧小鼠配对喂食的常氧小鼠,3天后血糖未出现显著变化。低氧暴露1周后,小鼠的低血糖效应显著加剧,而此时其急性应激反应(行为、摄食等)已恢复正常。此外,在所有检测的低氧时间点(1、2、3周),小鼠的葡萄糖耐量均得到显著改善。上述结果证实,单纯缺氧即可重现高海拔人群中观察到的葡萄糖耐量增强现象。
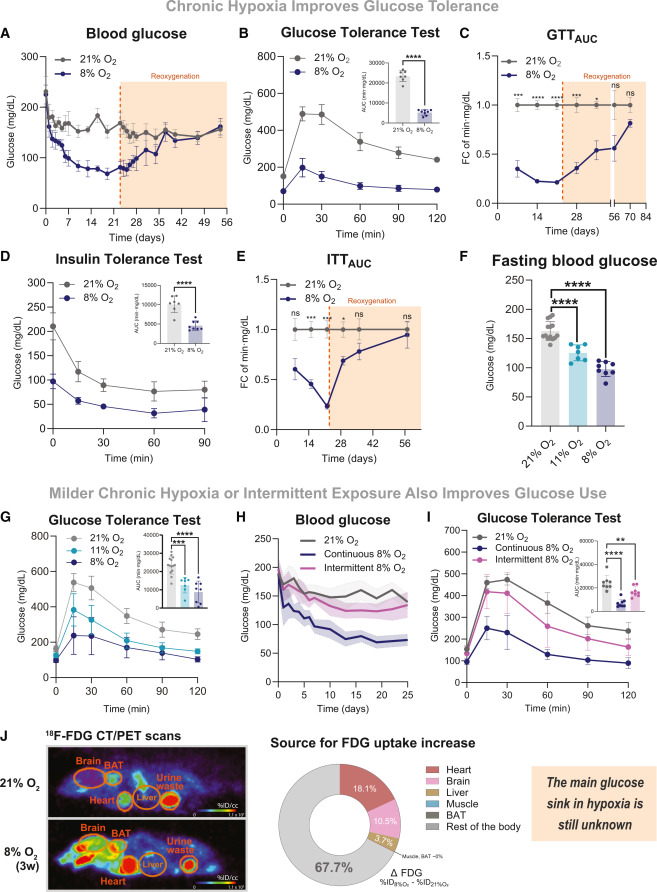
为进一步明确葡萄糖耐量改善的成因和动态变化,我们将适应低氧的小鼠恢复至常氧环境,定期监测血糖和体重,以确定血糖恢复正常的时间。结果显示,恢复常氧14天后,小鼠基础血糖恢复至常氧水平,而葡萄糖耐量在恢复常氧1个多月后才完全恢复正常。这表明,介导缺氧下葡萄糖耐量改善的机制,无需持续的缺氧暴露,且在缺氧停止后仍能维持数周。
为探究葡萄糖耐量改善是否依赖胰岛素,我们在小鼠缺氧和复氧阶段进行了胰岛素耐量实验。结果发现,缺氧时小鼠的胰岛素敏感性下降,且随复氧逐步恢复,其恢复速度快于葡萄糖耐量。由于低血糖会诱导机体的代偿性调节反应,这种胰岛素效应的降低可能反映了机体对持续性低血糖的全身适应性变化。该发现表明,缺氧下葡萄糖耐量的改善独立于胰岛素信号的增强,与此前研究中“慢性缺氧会降低循环胰岛素水平”的结论一致。因此,慢性缺氧下葡萄糖清除能力的提升是胰岛素非依赖性的。
为验证轻度缺氧干预是否也能改善葡萄糖耐量,我们分析了**轻度低氧方案**和**间歇性低氧**对小鼠葡萄糖稳态的影响。在轻度低氧实验中,将小鼠分别置于常氧(21% O₂)、中度低氧(11% O₂)和重度低氧(8% O₂)环境1周,检测其空腹血糖和葡萄糖耐量。有趣的是,中度低氧(11% O₂)即可显著降低小鼠空腹血糖,并大幅改善葡萄糖耐量。值得注意的是,空腹血糖和葡萄糖耐量均表现出氧浓度依赖性效应,而缺氧适应程度本身由氧供水平决定,这两种低氧方案诱导的红细胞压积升高也印证了这一点。低血糖的程度与红细胞压积的增加呈直接相关性,表明本研究的实验模型可推广至轻度缺氧条件。
在间歇性低氧实验中,将8周龄野生型雄性小鼠分别置于持续常氧(21% O₂)、持续低氧(8% O₂)或常氧-低氧交替的间歇性低氧环境25天。间歇性低氧方案为:小鼠睡眠期暴露于8% O₂低氧环境8小时,活动期恢复至21% O₂常氧环境16小时。结果显示,间歇性低氧可适度降低小鼠基础血糖,且不影响体重,表明小鼠在低氧环境中睡眠并未导致摄食受损。同样,间歇性低氧下小鼠的体温未出现急性降低,甚至在慢性暴露后有升高趋势。在干预第7、14、25天进行的葡萄糖耐量实验表明,小鼠的葡萄糖耐量在整个干预期间均得到适度且持续的改善。综上,即使是更温和、更具临床转化潜力的缺氧干预手段(如间歇性低氧疗法),也能适度增强葡萄糖耐量。
截至目前,所有数据均表明葡萄糖清除能力增强是缺氧性低血糖的主要成因,为验证该结论,我们评估了葡萄糖生成途径在该现象中的作用。通过丙酮酸耐量实验探究糖异生的作用,结果显示,肝脏糖异生对缺氧下的血糖降低无显著贡献。综上,葡萄糖清除能力增强是缺氧下葡萄糖耐量改善的最合理解释。
本研究前期的血糖检测均采用手持血糖仪,考虑到部分血糖仪在不同红细胞压积范围内检测时存在技术偏差,我们采用液相色谱-质谱联用技术(LC-MS),单独分析血浆组分的血糖水平作为验证方法。结果显示,低氧(8% O₂)暴露3周的小鼠,其血浆葡萄糖浓度较常氧小鼠降低35%,与此前血糖仪检测全血得到的降低幅度高度一致。此外,我们利用配对的常氧和低氧样本,详细对比了LC-MS检测血浆血糖与血糖仪检测全血/血浆血糖的结果,发现不同氧浓度下两种检测方法的结果具有高度相关性。值得注意的是,血糖仪检测血浆血糖的准确性极高,因此本研究将该方法作为血糖定量的金标准。上述结果验证了本研究血糖检测的准确性,也支持了实验方法的可靠性。
缺氧下葡萄糖耐量改善并非由内脏器官葡萄糖摄取增加导致,提示存在未知葡萄糖汇
为明确缺氧条件下葡萄糖清除的主要部位,我们分析了此前完成的正电子发射断层扫描/计算机断层扫描(PET/CT)成像数据,利用示踪剂2-脱氧-2-[¹⁸F]氟-D-葡萄糖(FDG)检测葡萄糖摄取。向常氧(21% O₂)或低氧(8% O₂)暴露3周的小鼠注射¹⁸F-FDG,以放射性¹⁸F-FDG的细胞摄取量作为葡萄糖摄取的替代指标。为评估缺氧暴露后葡萄糖摄取发生变化的主要器官的贡献,我们定量分析了这些器官及小鼠全身在缺氧下¹⁸F-FDG累积量的总增加量。令人意外的是,内脏器官并非缺氧下葡萄糖摄取增加的主要部位,约70%的增加量无法用内脏器官解释。该结果表明,介导缺氧下葡萄糖消耗增加的主要葡萄糖汇尚未被发现,也促使我们扩大研究范围。
红细胞增多是改善血糖控制的必要且充分条件
红细胞是人体数量最多的细胞类型,小鼠在8% O₂低氧环境中适应4周后,红细胞总质量近乎翻倍,该现象被称为红细胞增多症。由于红细胞缺乏线粒体,其葡萄糖代谢主要通过糖酵解途径和磷酸戊糖途径进行。已有研究表明,缺氧条件下红细胞的糖酵解通量显著高于磷酸戊糖途径,两者的相对通量分别约为90%和10%。据此我们提出假说:缺氧下红细胞数量增加与单个红细胞糖酵解增强的协同作用,可能是缺氧条件下葡萄糖摄取增加的主要原因。
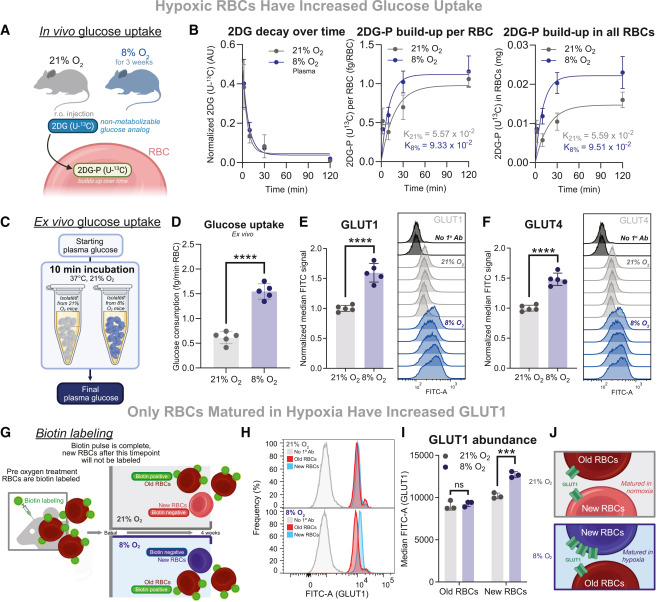
为直接验证缺氧时红细胞是否为主要葡萄糖汇,我们通过多次放血逆转低氧小鼠的红细胞增多症,探究该干预是否能缓解小鼠的血糖降低。具体而言,对置于常氧或低氧环境的8周龄雄性小鼠,每3天抽取其总血容量的15%,持续4周;同时设置未放血的常氧和低氧对照小鼠,对所有小鼠进行血液学和血浆血糖分析。结果显示,放血后的低氧小鼠,其红细胞压积恢复至与常氧对照小鼠相当的水平,且放血未显著改变其他血液指标。重要的是,红细胞压积恢复正常后,未放血低氧小鼠的缺氧性低血糖也显著缓解,血糖稳定在约170 mg/dL。此外,低氧暴露4周后的葡萄糖耐量实验表明,放血可显著逆转低氧小鼠的葡萄糖耐量改善效应。放血与未放血的低氧小鼠,其外周组织的葡萄糖转运蛋白表达无显著差异,排除了其他器官在该效应中的作用。综上,这些数据证实,耗竭红细胞可使低氧小鼠的血糖基本恢复正常,强烈提示红细胞增多症是缺氧下葡萄糖耐量增强的**必要条件**。
为直接验证红细胞丰度增加是否**足以**诱导与缺氧条件下相当的低血糖,我们开展了红细胞输注实验。将常氧饲养的雄性小鼠分为三组,连续两天每天进行两次眶后注射,分别输注生理盐水、常氧供体小鼠的红细胞或低氧供体小鼠的红细胞;在最后一次输注1天后,对所有小鼠进行血液学和血浆血糖检测。结果显示,与生理盐水处理组相比,红细胞输注组小鼠的红细胞数量显著增加,且增加幅度与低氧暴露2周的小鼠相近。正如预期,部分红细胞指标(如平均红细胞体积)因供体氧环境不同存在轻微差异,反映了常氧和低氧红细胞的固有区别,而其他血液学指标无显著变化。两个红细胞输注组均表现出明显的低血糖,且输注低氧红细胞的小鼠低血糖效应略强。输注常氧或低氧红细胞的小鼠,其终点血浆葡萄糖浓度均显著降低,与低氧暴露2周后的降低幅度相当。红细胞输注组与生理盐水注射组小鼠的外周组织葡萄糖转运蛋白表达无显著差异。综上,这些发现证实,红细胞数量升高**足以**诱导低血糖,支持“缺氧条件下红细胞作为主要葡萄糖汇”的假说。
缺氧小鼠的红细胞葡萄糖摄取能力增强
我们进一步探究,缺氧下的降血糖效应是否完全由红细胞数量增加导致,或缺氧是否还能增强单个红细胞的葡萄糖摄取能力。为解答该问题,我们以稳定同位素葡萄糖类似物——全碳13标记的2-脱氧-D-葡萄糖([U-¹³C]2DG)为示踪剂开展实验。与¹⁸F-FDG类似,[U-¹³C]2DG可通过葡萄糖转运蛋白进入细胞,磷酸化后被滞留于细胞内,生成[U-¹³C]2DG-6-磷酸,且该分子无法进一步参与糖酵解,因此其累积量可作为葡萄糖摄取的替代指标。向常氧或低氧暴露3周的小鼠眶后注射[U-¹³C]2DG,在注射后2、10、30和120分钟采集血液,通过LC-MS检测血浆[U-¹³C]2DG浓度。结果显示,两组小鼠的血浆[U-¹³C]2DG衰减动力学相似,表明示踪剂从血浆中的清除速率相当;但低氧小鼠红细胞内的[U-¹³C]2DG-6-磷酸累积速度显著更快,反映出单个红细胞的葡萄糖摄取能力增强。若考虑到缺氧下红细胞池的扩增,该效应则更为显著。上述结果证实,缺氧可在体内增强单个红细胞的葡萄糖摄取能力,进一步促进缺氧暴露下葡萄糖耐量的改善。
我们采用体外实验作为另一验证方法,进一步评估红细胞的葡萄糖摄取能力。收集常氧或低氧暴露4周小鼠的血液,通过离心分离红细胞;将等体积的常氧或低氧小鼠红细胞,与等体积血浆按1:1比例孵育,并将初始葡萄糖浓度标准化为一致。在37℃常氧条件下孵育10分钟后,检测血浆中残留的葡萄糖浓度,以计算葡萄糖摄取量;同时进行血液学分析,准确定量每个样本的红细胞数量。结果显示,低氧小鼠的红细胞,其单个细胞的葡萄糖摄取量较常氧对照增加2.5倍。重要的是,该实验全程在常氧条件下进行,因此低氧来源红细胞的葡萄糖摄取增强,反映的是一种持续性的功能适应,而非对低氧的超急性应答。综上,体外实验数据进一步证实,慢性缺氧可诱导单个红细胞的葡萄糖摄取能力产生长效增强。
为探究缺氧下单个红细胞葡萄糖摄取增加是否由红细胞内葡萄糖转运蛋白上调介导,我们采用流式细胞术分析其蛋白表达量。在成熟红细胞中,葡萄糖转运蛋白1(GLUT1)是主要的葡萄糖转运载体,而在红细胞生成增加的阶段,葡萄糖转运蛋白4(GLUT4)的表达也可能上调。因此,我们分析了常氧或低氧暴露3周小鼠红细胞中GLUT1和GLUT4的单个细胞蛋白表达量,同时还纳入了糖酵解关键酶己糖激酶I(HK1)和甘油醛-3-磷酸脱氢酶(GAPDH)进行分析。结果显示,缺氧暴露使红细胞中GLUT1和GLUT4的表达量分别显著上调60%和48%,其表达分布宽度增加,提示缺氧下红细胞的异质性增强。值得注意的是,低氧红细胞中GAPDH的蛋白表达量也有所增加,而HK1表达无显著变化。综上,这些结果表明,低氧红细胞的葡萄糖摄取增强,可能由这些葡萄糖转运蛋白的表达上调驱动。
由于成熟红细胞无法合成新蛋白,我们推测缺氧下红细胞生成增强,可能是葡萄糖转运蛋白上调的原因。为验证该推测,我们开展了生物素示踪实验:连续3天向小鼠注射生物素,标记所有现存的红细胞;随后将小鼠置于常氧或低氧环境,使未被标记的新生红细胞成熟。低氧暴露4周后,分析小鼠生物素阳性(“旧”红细胞)和生物素阴性(“新”红细胞)群体中GLUT1的表达量,发现GLUT1的上调特异性发生在新生红细胞中。因此,慢性低氧条件下,新生成熟红细胞至少在一定程度上介导了低氧红细胞葡萄糖摄取的上调。
缺氧增强红细胞糖酵解通量,促进血红蛋白变构调节因子2,3-DPG的合成
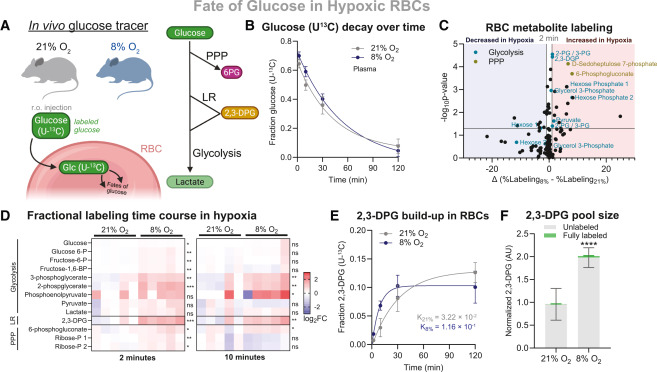
我们接下来探究了红细胞葡萄糖摄取增强后的代谢去向,以全碳13标记的D-葡萄糖([U-¹³C]葡萄糖)为示踪剂开展体内示踪实验。向常氧或低氧暴露3周的小鼠眶后注射[U-¹³C]葡萄糖,在注射后2、10、30和120分钟采集血液,通过LC-MS分析血浆[U-¹³C]葡萄糖的时间变化规律及红细胞代谢物。正如预期,血浆中的标记葡萄糖快速衰减,提示细胞对葡萄糖的快速清除。为全面分析缺氧条件下红细胞内葡萄糖的代谢去向,我们开展了半靶向代谢组学分析,结果显示,低氧小鼠红细胞中,LR支路的主要产物2,3-DPG及糖酵解中间产物的标记率增加速度显著更快,与此前研究结果一致;部分由葡萄糖标记的代谢物(如1,6-二磷酸己糖)在缺氧下标记率升高,而另一些代谢物(如己糖,包括葡萄糖和己糖异构体)的标记率则降低。
对糖酵解、LR支路和磷酸戊糖途径中间产物的靶向定量分析证实,标记的2,3-DPG与其他糖酵解中间产物均显著累积。重要的是,2,3-DPG作为血红蛋白的关键变构调节因子,可促进缺氧组织的氧释放。时间依赖性的标记率分析显示,低氧红细胞中2,3-DPG的标记速率常数较常氧红细胞增加约3.5倍。尽管精确的通量计算需要绝对池大小定量和非扰动性输注,但低氧小鼠红细胞中2,3-DPG的相对池大小和标记率均显著更高,表明葡萄糖向LR支路的代谢通量定性增加。综上,本研究数据证实,缺氧可显著增强红细胞在体内的葡萄糖利用,并将其导向血红蛋白调节因子2,3-DPG及其他糖酵解中间产物的合成。
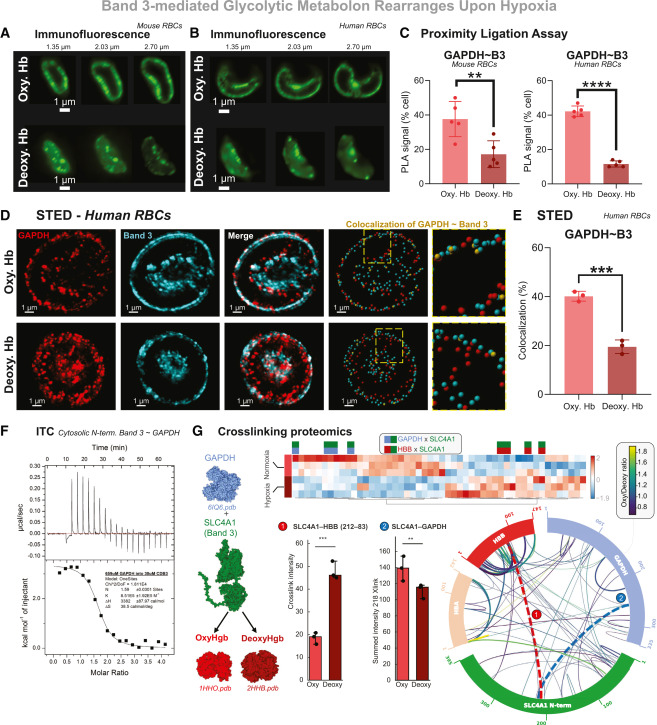
缺氧重构红细胞特异性糖酵解代谢体,促进糖酵解进程
为探究氧分压如何调控红细胞的葡萄糖代谢,我们验证了红细胞是否会发生氧依赖性的急性代谢重编程——这一机制可解释红细胞在缺乏转录和翻译机制的情况下,糖酵解仍能被快速激活的现象。已有研究提出,常氧条件下,糖酵解酶会通过结合红细胞膜上最丰富的整合蛋白——带3蛋白的N端,聚集在质膜上并处于失活状态;而缺氧时,脱氧血红蛋白会竞争性取代这种结合,使GAPDH等糖酵解酶释放至细胞质中,进而促进糖酵解通量。为在本研究体系中验证该机制,我们对氧合和脱氧状态的红细胞进行GAPDH免疫荧光染色,结果显示,在小鼠和人类红细胞中,GAPDH在常氧下均定位于质膜,而在缺氧下则重新分布至细胞质。
我们进一步采用邻近连接实验(PLA)检测GAPDH与带3蛋白的结合动力学,发现缺氧时两种物种红细胞中GAPDH与带3蛋白的相互作用均显著减少。此外,为更高空间分辨率地分析两者的共定位差异,我们对氧合和脱氧的人类红细胞进行受激发射损耗(STED)显微镜分析,发现缺氧条件下GAPDH与带3蛋白的共定位显著降低。
为直接探究两者的相互作用,我们以重组人源GAPDH和带3蛋白的N端胞质结构域(1-395位氨基酸)为研究对象,开展等温滴定量热法(ITC)实验,结果显示GAPDH可与该结构域紧密结合(解离常数Kd=1.18 μM)。值得注意的是,该亲和力高于此前报道的GAPDH与带3蛋白N端56个氨基酸短肽的结合力,提示除该固有无序的N端极端区域外,其他氨基酸残基也参与了结合过程。
为进一步探究该相互作用的结构基础,我们开展了基于定量串联质量标签(TMT)标记的交联蛋白质组学实验:将重组GAPDH与带3蛋白1-395位氨基酸片段共同孵育,并分别加入氧合或脱氧血红蛋白。结果显示,脱氧血红蛋白(尤其是83位残基的β珠蛋白)与带3蛋白212位残基的相互作用在缺氧下增强2.4倍,该相互作用会取代GAPDH与带3蛋白218位残基的结合。该分析进一步拓展了对带3蛋白与GAPDH相互作用的理解:带3蛋白的33位和40位残基分别与GAPDH的107位和84位残基交联,而带3蛋白的122、166、168、205、218和254位残基则与GAPDH的106、172、189、254和256位残基相互作用;这些区域均靠近GAPDH的活性位点(152位半胱氨酸和179位组氨酸),为带3蛋白结合抑制GAPDH活性提供了结构学解释。
综上,这些数据证实了红细胞糖酵解代谢体的氧依赖性重构:常氧下,GAPDH通过与带3蛋白结合被封存于质膜,抑制糖酵解通量;缺氧下,脱氧血红蛋白竞争性结合带3蛋白,使GAPDH释放至细胞质,从而激活糖酵解。该保守机制在小鼠和人类红细胞中均存在,解释了低氧条件下红细胞的急性代谢转换。
缺氧和红细胞输注可改善1型和2型糖尿病模型小鼠的高血糖症状
为探究缺氧诱导的红细胞数量增加和代谢重编程,是否可作为高血糖的治疗手段,我们验证了缺氧和红细胞压积升高在治疗1型和2型糖尿病中的有效性。
首先,我们评估了缺氧作为1型糖尿病高血糖治疗手段的潜力。通过向8周龄雄性小鼠腹腔注射链脲佐菌素(STZ)或生理盐水,构建1型糖尿病模型:STZ会破坏小鼠胰腺中分泌胰岛素的β细胞,导致胰岛素缺乏和高血糖。为更贴合临床病程,我们在STZ注射2周后,待小鼠明确出现高血糖后,再启动缺氧治疗。将STZ处理和生理盐水处理的小鼠随机分为常氧或低氧治疗组,持续3周。结果显示,低氧暴露使STZ处理和生理盐水处理的小鼠均出现红细胞增多症,而常氧条件下小鼠的红细胞数量无显著变化。在整个研究期间,每3天监测小鼠的基础血糖和体重,发现低氧处理的STZ小鼠,其初始升高的血糖显著降低;3周后的葡萄糖耐量实验表明,缺氧可完全逆转STZ诱导的葡萄糖耐量受损。该结果凸显了该效应的胰岛素非依赖性,也证实了该干预手段的治疗潜力。
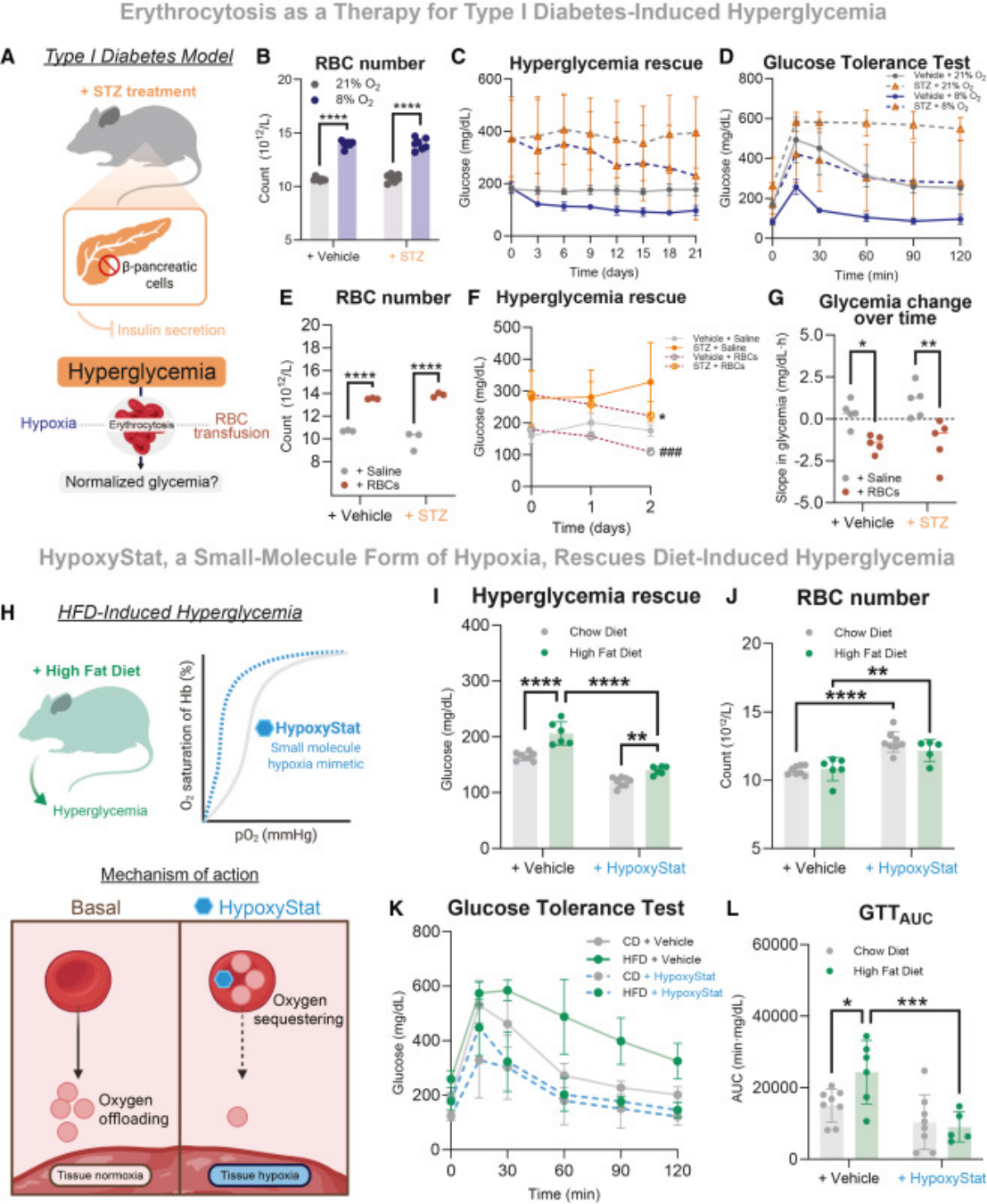
其次,为排除缺氧治疗的潜在多效性,我们探究了输注低氧红细胞是否足以改善糖尿病小鼠的高血糖。对生理盐水和STZ处理的小鼠,连续两天输注低氧红细胞或等体积生理盐水,纵向评估其血糖水平。结果显示,与生理盐水注射组相,红细胞输注组小鼠的红细胞数量显著增加;所有输注红细胞的小鼠(包括STZ处理的糖尿病小鼠)血糖均出现下降,证实红细胞数量增加足以改善1型糖尿病背景下的高血糖。
最后,我们评估了本研究团队近期研发的小分子缺氧模拟物HypoxyStat,是否能有效逆转高脂饮食(HFD)诱导的高血糖。HypoxyStat可提高血红蛋白的氧亲和力,抑制其向组织释放氧气,进而诱导局部组织缺氧。将饲喂普通饮食(CD)或高脂饮食的雄性小鼠,在常氧环境下每日灌胃给予生理盐水或HypoxyStat,持续2.5周。正如预期,与普通饮食对照组相,生理盐水处理的高脂饮食小鼠出现明显的高血糖;而HypoxyStat处理可完全消除高脂饮食诱导的高血糖,同时伴随显著的红细胞增多症,且未影响其他血液学指标。生理盐水处理的高脂饮食小鼠,其葡萄糖耐量显著受损,而HypoxyStat处理可使小鼠的葡萄糖耐量完全恢复正常,且该效应与饮食脂肪含量无关。综上,这些发现证实,本研究研发的小分子缺氧模拟物HypoxyStat,可有效改善饮食诱导肥胖相关的高血糖和葡萄糖耐量受损。
研究讨论
高海拔人群的血糖降低、葡萄糖耐量改善现象已被长期观察,且该特征在多种生物中均存在,但其背后的生理机制始终不明确。本研究证实,缺氧条件下红细胞是机体的主要葡萄糖汇。缺氧可显著增加红细胞数量,且新生红细胞的葡萄糖摄取能力增强;耗竭低氧小鼠的红细胞可使其血糖恢复正常,而在常氧下人工增加红细胞数量则足以诱导低血糖。红细胞数量增加,可能是解释慢性缺氧下葡萄糖清除能力增强的主要机制。此外,本研究还发现,缺氧可增强单个红细胞的葡萄糖摄取能力,这一效应至少部分由新生红细胞中GLUT1和GLUT4的蛋白表达上调介导。葡萄糖摄取的增加,可促进血红蛋白变构调节因子2,3-DPG的快速累积,而2,3-DPG是缺氧适应中氧释放的关键分子。这些发现揭示了一种此前未被发现的生理机制,也为高血糖的治疗提供了创新思路。
本研究证实,慢性缺氧下年轻红细胞的葡萄糖摄取能力增强,且伴随单个红细胞中GLUT1和GLUT4蛋白表达量的升高。GLUT1是成熟红细胞的主要葡萄糖转运载体,也有研究表明红细胞中存在GLUT4、GLUT3等其他葡萄糖转运蛋白的表达。由于成熟红细胞无细胞核,无法合成新蛋白,其葡萄糖摄取能力主要由发育早期的因素决定,包括造血祖细胞阶段确立的GLUT1和GLUT4表达水平。已知红系祖细胞中GLUT1的表达受HIF通路调控,而该通路在红细胞生成过程中及慢性缺氧条件下会被激活。由于红细胞中葡萄糖转运蛋白的半衰期接近红细胞自身的寿命,缺氧下观察到的葡萄糖摄取增强,可能主要归因于年轻红细胞池的扩增——这些红细胞在生成过程中建立了更高的葡萄糖转运蛋白表达水平,并保留了该特征。这与“高海拔反复登高后,机体的红细胞代谢适应会长期保留”的观点一致。未来研究将进一步剖析红细胞年龄和HIF通路对GLUT1、GLUT4表达的调控作用,以明确其在全身缺氧下血糖改善中的具体贡献。
除葡萄糖摄取增强外,本研究还观察到,缺氧时葡萄糖向2,3-DPG及其他糖酵解中间产物的转化显著加速。这些急性应答在数分钟内即可发生,而非数天/数周,因此无法用缺氧下新生红细胞的从头生成和转录重编程解释。在此背景下,带3蛋白介导的糖酵解酶动态区室化,可能是该效应的关键调控机制。常氧条件下,带3蛋白(红细胞质膜上的丰度蛋白)可结合并封存糖酵解酶,进而促进葡萄糖通过磷酸戊糖途径代谢,以合成烟酰胺腺嘌呤二核苷酸磷酸(NADPH),维持抗氧化防御系统;而缺氧时,脱氧血红蛋白使糖酵解酶从带3蛋白上解离,进而增强糖酵解通量,促进2,3-DPG累积。本研究证实,该机制在小鼠实验体系中成立,且在人类中高度保守。
本研究结果强烈支持“缺氧性低血糖主要由红细胞的葡萄糖摄取加速和增强导致”的结论,但糖异生、糖原分解或葡萄糖的吸收与分泌变化,也可能参与了全身低血糖的发生。丙酮酸耐量实验(肝脏糖异生能力的替代指标)的初步数据显示,不同氧环境下小鼠的糖异生能力无显著差异,但仍需进一步探究糖原分解、肠道葡萄糖吸收等其他途径,以全面理解缺氧对葡萄糖代谢的影响,而非仅局限于本研究观察到的葡萄糖摄取效应。
本研究数据也为人群水平的观察结果提供了新的解读,解释了此前高海拔居民血糖控制能力的未知差异。大多数高海拔人群的葡萄糖耐量均得到改善,但夏尔巴人是一个显著的例外——尽管长期暴露于高海拔环境,其血糖控制能力并未提升。从遗传学角度,夏尔巴人携带HIF2(EPAS1)及相关基因的缺氧适应性突变,可抑制缺氧诱导的红细胞生成,因此其红细胞压积水平显著低于其他高海拔人群;类似地,安第斯人群中携带EPAS1/HIF2错义突变rs570553380的个体,红细胞压积也较低。本研究为该现象提供了机制解释:夏尔巴人缺乏新生、高葡萄糖摄取能力的红细胞池扩增,因此无法获得其他高海拔人群典型的血糖获益。
本研究的发现不仅适用于高海拔人群,还具有更广泛的临床意义。例如,楚瓦什红细胞增多症患者在基础状态下,因HIF信号持续激活表现出显著的红细胞增多,且其血糖水平降低,这与本研究结果一致;促红细胞生成素(EPO)治疗在人类和动物模型中,均被证实可改善葡萄糖耐量、降低血糖;性腺功能减退男性的睾酮替代治疗,或性别确认治疗中的睾酮使用,可诱导红细胞增多,同时伴随胰岛素敏感性提升和高血糖恢复正常。相反,贫血患者常表现出高血糖,且糖尿病与贫血的发生密切相关,这些发现进一步支持了本研究提出的生理机制。综上,这些零散但一致的观察结果表明,红细胞在血糖控制中的作用长期被忽视,且可能具有更广泛的临床意义。
未来高血糖的治疗方向,可考虑使用本研究研发的小分子缺氧模拟物HypoxyStat,或输注经低氧保存的红细胞。但对于糖尿病这类相对可控的疾病,红细胞数量增加带来的血液黏度升高风险,可能抵消其治疗获益。因此,研发能促进红细胞快速更新、使红细胞池向年轻、高葡萄糖摄取能力的红细胞倾斜的治疗手段,可能是糖尿病管理的新方向——该干预手段不会影响血液黏度,且有望改善血糖控制。尽管如此,仍需开展更多研究,将本研究发现安全、有效地转化为临床治疗方案。
综上,本研究证实,缺氧条件下红细胞是全身葡萄糖代谢的关键调控因子。值得注意的是,Scherer等人近期发表的相关研究结果,进一步验证了本研究的观察结论。红细胞丰度增加和代谢重编程可显著增强葡萄糖摄取,使红细胞成为移动的葡萄糖储库。该发现解释了高海拔人群血糖控制改善的长期观察结果,也阐明了红细胞增多症和贫血患者中红细胞数量与血糖的临床关联。本研究为高血糖及相关代谢疾病的治疗开辟了新途径,包括调控红细胞动态变化,以及使用HypoxyStat等小分子缺氧模拟物进行治疗。
研究局限性
本研究存在以下几点局限性:第一,所有小鼠实验均采用C57BL/6J品系,该品系小鼠的葡萄糖耐量较其他品系更低。尽管将研究结果拓展至多种小鼠品系会更理想,但该现象在人类和其他物种中均存在,表明其潜在机制不具有品系特异性,且在哺乳动物中高度保守。第二,尽管流行病学数据显示,不同性别和年龄人群的血糖控制改善效应相当,但为保证实验一致性,本研究所有实验均仅使用年轻雄性小鼠。考虑到衰老过程中红细胞生成显著减少,且不同性别的红细胞群体动态存在已知差异,未来有必要深入分析年龄和性别在红细胞调控葡萄糖稳态能力中的作用。第三,本研究证实葡萄糖转运蛋白的上调特异性发生在“缺氧环境中生成的红细胞”中,但尚未明确该现象的分子机制。未来研究将剖析红系祖细胞中的信号通路,以明确缺氧下葡萄糖转运蛋白上调的具体机制。
转载本文请联系原作者获取授权,同时请注明本文来自孙学军科学网博客。
链接地址:https://wap.sciencenet.cn/blog-41174-1522799.html?mobile=1
收藏