
 精选
精选
癌症“过往”的表观遗传线索可预测其“未来”
一种通过动态DNA修饰模式追踪肿瘤演化的创新方法,有望应用于临床以预测癌症进展。
论文概要
- 癌症是一种动态疾病,但通过单一肿瘤样本的基因组分析追踪其演化过程存在较大挑战。
- 基因组中的特定位点会发生甲基化这一表观遗传修饰,其中部分位点的甲基化与去甲基化状态会随时间随机波动。
- 癌细胞群体中这些位点的甲基化模式会形成独特的“条形码”,可用于追溯该细胞群体的生长历史。
- 在发表于《自然》(*Nature*)的论文中,加布特(Gabbutt)等人[1]提出一种利用这种甲基化条形码推断血液系统癌症生长动态及临床结局的方法。
帕夫洛·卢齐克(PAVLO LUTSIK):通过甲基化追溯肿瘤历史
癌症的发展可能需要数年甚至数十年时间。在此期间,肿瘤会经历一系列复杂的演化事件,其标志是恶性细胞谱系(克隆)间的竞争——这些克隆会逐渐增强生存与增殖能力,这一过程被称为“选择与扩张”。克隆获得的特征不仅决定癌症患者的预后,也是理解如何实现癌症早诊、阻断与预防的关键。
然而,要全面理解癌症发展,需要进行长期观察,但这在实际场景中几乎难以实现。研究人员通常只能获取少量样本,包括诊断性活检样本、手术切除的原发肿瘤样本,有时还包括少数转移灶样本。从这些有限的“快照”中重建复杂的肿瘤演化轨迹,是一项公认的难题。
由于大多数基因突变被认为是不可逆的,因此可用于定义肿瘤的克隆组成,目前推断肿瘤演化的主流方法是分析基因组数据。这类分析可基于 bulk 组织样本[2]、单细胞样本[3]的数据,近年来还可结合组织的空间分辨基因组图谱[4]。与大多不可逆的遗传改变不同,表观遗传改变(对DNA及其包装蛋白的化学修饰)大多具有可逆性。尽管某些表观遗传改变(如DNA甲基化的大规模丢失与局部获得)已知与恶性转化、疾病进展及演化相关[5,6],但其可逆性使得利用患者样本重建肿瘤演化轨迹的应用受限。
在2022年发表的一项开创性研究[7]中,加布特及其团队打破常规:他们发现部分DNA甲基化位点可作为克隆特异性分子条形码。在最新发表于《自然》的研究中,加布特等人[1]基于这一发现,提出了一个用于推断肿瘤演化的综合数学建模框架,并将其工具命名为EVOFLUx。
人类基因组中约有2800万个可发生甲基化的核苷酸位点,这些位点均为胞嘧啶后接鸟嘌呤的结构(即CpG位点)。在二倍体细胞(含两套完整染色体的细胞)中,大多数CpG位点在一对同源染色体上均有分布,且每个位点的甲基化状态呈二元特征——要么甲基化,要么去甲基化。加布特团队的核心发现是:一小部分特定的“动态CpG位点”(fCpG)会以特定速率随机切换甲基化状态,且两条染色体上的位点状态相互独立。若某一fCpG位点的两条染色体拷贝均甲基化,其甲基化“值”为1;若均未甲基化,值为0;若仅一条拷贝甲基化,值为0.5(图1a)。
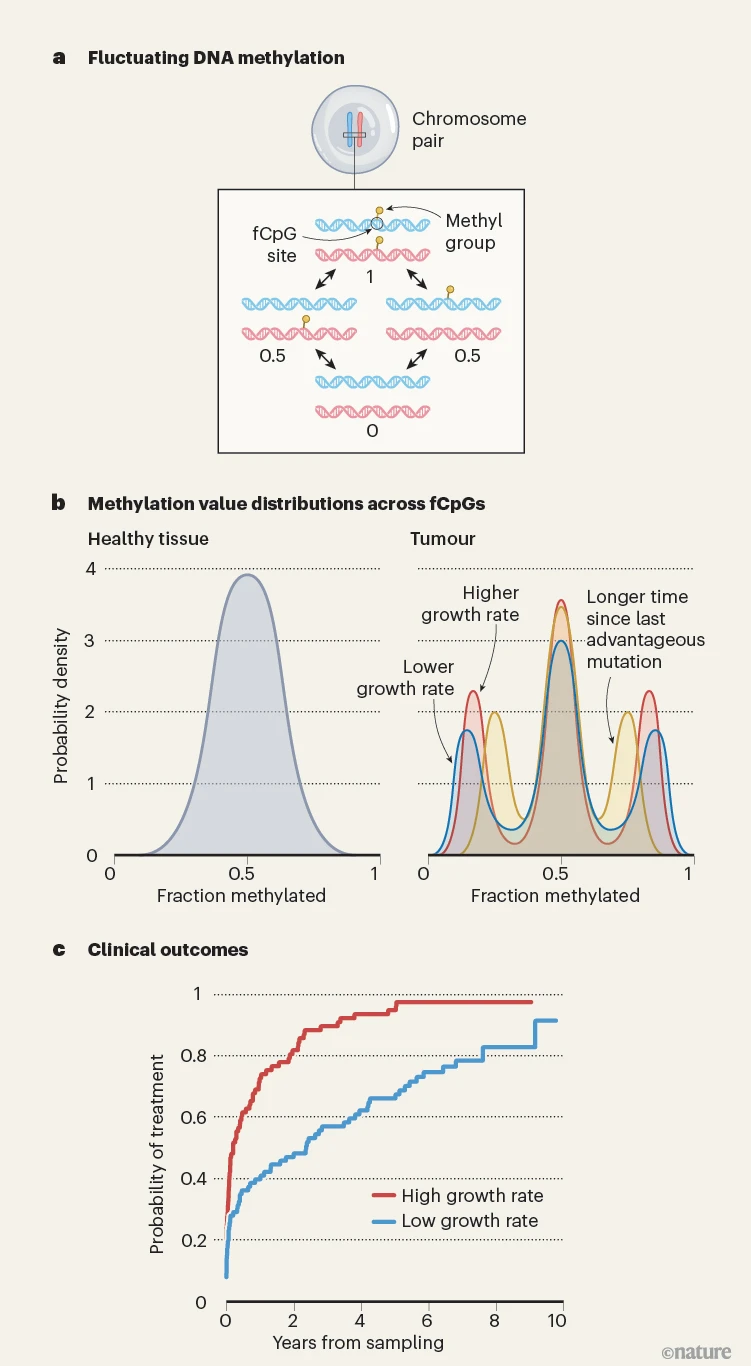
图1 | 追踪癌症演化的计算方法
a)基因组中被称为CpG位点(DNA序列中胞嘧啶后接鸟嘌呤)的区域可发生甲基化表观遗传修饰。加布特等人[1]发现,某些“动态CpG位点”(fCpG)会在甲基化与去甲基化状态间随机切换,且其两条染色体拷贝(同源染色体对中的每条染色体各含一个拷贝)的状态相互独立。因此,每个fCpG位点的甲基化值可为1、0.5或0。
b)在包含多个细胞谱系(克隆)的健康细胞群体中,单个组织样本内所有fCpG位点的甲基化值分布集中在0.5左右。在癌症中,部分克隆会获得优势突变,使其扩张速度远快于甲基化波动速度;此时,单个fCpG位点会被“锁定”在不同状态(0、0.5、1),而非维持在平均稳态(0.5),从而在接近0和1的位置形成新的峰值。作者设计并应用了名为EVOFLUx的数学框架,通过分布曲线的形态解读肿瘤生长参数,例如生长速率、自上次驱动克隆扩张的优势突变发生以来的时间。
c)这些参数与疾病进展的临床指标相关。例如,在慢性淋巴细胞白血病患者中,EVOFLUx归类为“高生长速率”的患者,其首次治疗时间(反映癌症自然进展的指标)短于“低生长速率”患者(改编自参考文献[1]的图5)。
在由多个克隆群体组成的健康组织中(每个克隆的细胞数量相近),大量克隆中任意fCpG位点的平均甲基化值均接近0.5。但当某个克隆获得足够多的优势突变,且扩张速度远超过fCpG位点的状态切换速度时,所有fCpG位点的甲基化模式会在其子代细胞中固定,形成独特的分子条形码——这一特征可在bulk数据中检测到。
最终,原本集中在0.5附近的fCpG甲基化单峰分布,会在接近0和1的位置出现额外峰形,分别对应完全去甲基化和完全甲基化的fCpG位点,形成“W”形多峰分布(图1b)。加布特等人证实,该分布曲线的具体形态取决于自上次克隆扩张以来的时间,以及肿瘤生长的关键参数。EVOFLUx采用贝叶斯建模这一统计技术,估算这些生长参数,并揭示了1976份淋巴系统恶性肿瘤(影响白细胞的癌症)样本的演化细节。
值得关注的是,EVOFLUx能从基于微珠的芯片数据中提取详细的回溯性信息——这类芯片可量化细胞群体的甲基化水平,但生成的是本质上的bulk数据,完全掩盖了潜在的细胞异质性。尽管这类平台无法解析单个DNA分子的甲基化信息,但其高度优化的性能与成本效益,使其有望快速转化为临床常规检测方法。
目前一个关键的未解决问题是:哪些分子机制导致了fCpG位点的动态特征?作者已尽力验证,这些波动与DNA序列本身的任何改变无关,即属于纯表观遗传过程。此外,有研究通过分析单细胞DNA甲基化的新方法[8],独立证实了克隆特异性CpG亚群的存在。这提示人类基因组中可能存在比加布特等人描述的更多类fCpG位点。要验证这一点并阐明其潜在分子机制,还需进一步研究。未来,针对EVOFLUx的改进需求将十分迫切——例如适配靶向长读长DNA测序、快速长读长DNA测序等最新高信息量检测技术,以在癌症演化评估中整合基因组与表观基因组事件。
韦塞林·马诺伊洛维奇(VESELIN MANOJLOVIC)& 乔治·S·瓦西利乌(GEORGE S. VASSILIOU):预测癌症结局的可推广方法
在临床相关尺度上重建癌症发展过程,长期以来都是一项重大挑战。在论文中,加布特等人[1]应用EVOFLUx框架——这是一种通过单一时间点获取的bulk DNA甲基化数据推断肿瘤演化历史的计算工具。该方法使复杂的演化分析能应用于可推广的临床场景,为个体肿瘤历史提供了详细见解——而在缺乏系列样本的情况下,这类分析此前难以实现。
通过对淋巴系统恶性肿瘤样本应用EVOFLUx,研究人员获得了多项具有临床意义的发现,例如急性与慢性恶性肿瘤在生长速率和群体规模上的显著差异,以及这些参数与临床结局的关联。慢性淋巴细胞白血病(CLL)是最常见的淋巴系统恶性肿瘤之一,研究发现其在大多数情况下呈“中性生长”(即无亚克隆群体扩张),但在EVOFLUx检测到亚克隆扩张的案例中,约30%存在选择与扩张过程。
针对两例进展为侵袭性淋巴瘤的慢性淋巴细胞白血病患者,作者通过系统发育分析发现:最终发生转化的克隆,在淋巴瘤出现临床症状前数十年就已与其他克隆分化。这提示这类高风险转化事件可能在数年前就可被检测到。此外,该模型推断的生长速率与慢性淋巴细胞白血病患者的临床结局高度相关:生长速率越高,首次治疗时间越短(图1c),总生存期越短。这种预后信息独立于已确立的标志物(如*TP53*基因突变、*IGHV*基因重排)。
该方法的一大显著优势在于,它依赖成本相对较低、普及率高的甲基化芯片,而非资源消耗大且技术变异性高的甲基化测序技术。这种可推广性使其有望整合到常规诊断中,用于疾病早筛或根据疾病风险对个体进行分层。
尽管该研究主要聚焦血液系统癌症,但其方法可扩展至实体瘤,并适配基于测序的技术。若结合终生获得性突变数据,可实现对肿瘤更全面的特征分析。此外,该方法还可改造用于分析循环肿瘤游离DNA(cfDNA),从而实现对多种癌症演化动态的无创实时监测。
EVOFLUx是一项重要突破,它不再将癌症视为静态疾病,而是还原其动态演化的本质。这一框架能将看似固定的甲基化图谱,转化为丰富的克隆变化历史记录。通过简单检测即可测量肿瘤生长速率、预测预后或潜在侵袭性转化的能力,为主动化、个体化癌症诊疗带来了巨大潜力。
转载本文请联系原作者获取授权,同时请注明本文来自孙学军科学网博客。
链接地址:https://wap.sciencenet.cn/blog-41174-1501744.html?mobile=1
收藏