
 精选
精选
睡眠压力的线粒体起源
为全面、无偏地了解可能构成睡眠需求基础的大脑分子变化,我们对来自休息状态和睡眠剥夺状态果蝇的单细胞转录组进行了表征。研究发现,在投射至背侧扇形体1、2(dFBNs)的睡眠控制神经元中(而非在全脑普遍),睡眠剥夺后上调的转录本几乎全部编码参与线粒体呼吸和ATP合成的蛋白质。这些基因表达变化伴随着线粒体碎片化、线粒体自噬增强,以及线粒体与内质网之间接触数量的增加,从而形成补充过氧化脂质⁵的通道3、⁴。这些形态学变化在恢复性睡眠后可逆转,且会因呼吸链中电子溢出⁶、⁷的产生而减弱。在dFBNs中诱导或阻止线粒体分裂或融合⁸⁻13,会以相反方向改变睡眠及睡眠控制细胞的电特性:线粒体过度融合会增加神经元兴奋性和睡眠量,而线粒体碎片化则会降低神经元兴奋性和睡眠量。强制清醒后,由于这些神经元在觉醒介导的抑制过程中ATP消耗减少1⁴,dFBNs中的ATP浓度会升高,这会增加其线粒体电子泄漏⁷。与此观点一致,使电子通量与ATP合成解偶联1⁵可缓解睡眠压力,而加剧电子供应与ATP需求之间的不匹配(通过利用光驱动质子泵为ATP合成供能1⁶)则会促使睡眠发生。睡眠或许如同衰老1⁷、1⁸一样,是有氧代谢不可避免的结果。
Mitochondrial origins of the pressure to sleep | Nature
睡眠压力作为睡眠稳态中的过程变量,尚缺乏物理层面的解释。尽管长时间清醒与大脑中的诸多变化相关——包括神经元放电模式1⁹、2⁰、突触连接强度21、亚细胞结构组织22⁻2⁴、代谢物浓度2⁵、2⁶以及代谢和基因表达程序23、2⁷、2⁸的改变——但总体而言,这些变化究竟是睡眠需求增加的原因还是结果,仍无法确定。或许,唯一能将因果关系与相关关系区分开的机会存在于那些在睡眠诱导和维持中发挥积极作用的特殊神经元中2⁹;在这些细胞中,睡眠的直接(或许也是最终)原因必定与调节神经元放电的过程直接关联。为尽可能全面且无偏地描绘这些过程的分子决定因素,我们收集了休息状态和睡眠剥夺状态果蝇大脑的单细胞转录组3⁰(扩展数据图1a)。一种可编码的荧光标记使我们能够识别并富集二十多个投射至中央复合体背侧扇形体1、2(dFBNs)的促睡眠神经元,并将它们对睡眠缺失的转录组反应与其他可识别细胞类型的反应进行比较。
线粒体和突触中睡眠缺失的痕迹
我们将果蝇大脑解离为单细胞悬液,并通过流式细胞术分离出在R23E10-GAL4(参考文献31)控制下表达GFP的神经元(扩展数据图1b)。我们对GFP阳性和GFP阴性组分中的细胞进行了单细胞RNA测序(scRNA-seq;10X Chromium);在获得的13,173个高质量细胞中(扩展数据图1c、d),R23E10-GAL4标记的神经元通过GFP、神经元标记物胚胎致死异常视觉蛋白(elav)、神经元突触小泡蛋白(nSyb)以及咽侧体抑制素A(AstA)受体1(AstA-R1)的表达来识别,其转录增强子提供了驱动R23E10-GAL4系表达的基因组片段。R23E10-GAL4域中的大多数神经元都含有合成和释放谷氨酸或γ-氨基丁酸(GABA)的机制(扩展数据图1e);有些还转录编码囊泡乙酰胆碱转运蛋白(VAChT)的基因(扩展数据图1e)——这在非胆碱能神经元中很常见,它们依靠微小RNA来抑制其翻译32。事实上,荧光传感器检测到dFBNs释放谷氨酸,但未检测到释放乙酰胆碱2。在果蝇中枢神经系统中,谷氨酸和GABA都作用于配体门控氯离子通道,这与已知的dFBNs对其突触后伙伴的抑制作用一致2、33。R23E10-GAL4模式中通过这两种抑制性递质中的一种进行信号传递的神经元在解剖学上是分离的2:一对GABA能细胞靶向食道下神经节,而更大的谷氨酸能群体支配背侧扇形体。后一组真正的dFBNs在大脑中所有谷氨酸能神经元中形成了一个包含323个细胞的独特基因表达簇(图1a和扩展数据图1f、g)。该簇由已知的dFBN增强子控制的基因(扩展数据图1h)以及编码已知dFBN标记物的基因定义,例如Rho GTP酶激活蛋白31无横脉-c、多巴胺受体1⁴Dop1R2、血清素受体3⁴5-HT2B和神经肽33AstA(图1b),但某些标记物的分布表明存在进一步的细分。例如,AstA仅限于dFBNs的一个子集(图1b),这与在整个dFBN群体中通过RNA干扰(RNAi)抑制其表达后出现的相对轻微的睡眠表型一致33。
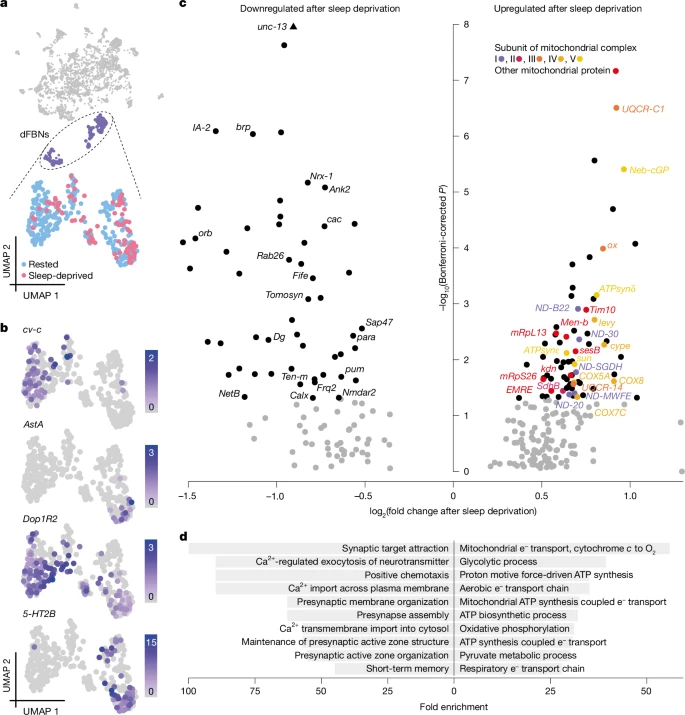
图1:dFBNs对睡眠剥夺的转录反应
a. 根据基因表达谱,谷氨酸能神经元(灰色)的均匀流形近似和投影(UMAP)表示。dFBNs(紫色)形成一个独特的簇,包含来自休息状态(蓝色,n = 237个细胞)和睡眠剥夺状态大脑(红色,n = 86个细胞)的细胞。b. dFBN标记物的对数标准化表达水平。c. dFBNs中睡眠历史依赖性基因表达变化的火山图。Bonferroni校正后P < 0.05(双侧Wilcoxon秩和检验)的信号以黑色表示;标签标识定位于突触或线粒体的蛋白质产物;颜色表示线粒体呼吸复合物的亚基。unc-13的P值超过y轴限制,绘制在图表顶部。d. 在差异表达的dFBN基因集中,上调(右)和下调(左)的前10个“生物学过程”基因本体术语的富集情况。
对12小时夜间睡眠剥夺后dFBNs中水平发生变化的122个转录本进行的基因本体分析显示,仅存在两个与睡眠需求相关的过程:线粒体能量代谢和突触传递(图1c、d和扩展数据图2a-e)。睡眠缺失导致编码电子传递复合物I-IV、ATP合酶(复合物V)、ATP-ADP载体sesB以及三羧酸循环酶(柠檬酸合酶kdn、琥珀酸脱氢酶B亚基和苹果酸脱氢酶Men-b)成分的转录本选择性上调,而参与突触组装、突触囊泡释放和突触前稳态可塑性3⁵的基因产物则选择性下调(图1c)。在我们的分析实际范围内(无法涵盖所有可能的神经元类型),这种睡眠缺失的转录组特征似乎是dFBNs所特有的:在我们数据中具有可比数量代表性的两个细胞群中——即触角叶的投射神经元(317个细胞)(扩展数据图3a-d)和蘑菇体的肯扬细胞(603个细胞)(扩展数据图3e-h)——均未发现该特征,并且在对所有12,850个非dFBN细胞的综合分析中也未检测到(扩展数据图3i、j)。然而,一项独立的转录组研究提供了微妙的线索,表明睡眠历史不仅可能改变dFBNs中的线粒体成分水平,还可能改变椭球体R5神经元(睡眠稳态的另一个组成部分)中的线粒体成分水平2⁸。本文的其余部分探讨了dFBNs中编码线粒体蛋白的基因差异表达的原因和结果;一项平行研究2探讨了突触前可塑性在更广泛的睡眠需求依赖性dFBN动态中的作用。
线粒体电子过剩诱导睡眠
线粒体成分在dFBNs对睡眠剥夺的转录反应中占据显著地位(图1c、d和扩展数据图2),这为睡眠与有氧代谢存在根本联系的假设提供了无偏支持。随着发现dFBNs通过将其促睡眠尖峰放电与线粒体呼吸相连接的机制来调节睡眠⁷,这一假设获得了坚实的机制基础。该机制的核心是Hyperkinetic,即电压门控钾通道Shaker的β亚基,它调节dFBNs的电活动⁷、1⁴。Hyperkinetic是一种不寻常的醛酮还原酶,具有稳定结合的烟酰胺腺嘌呤二核苷酸磷酸辅因子,其氧化状态(NADPH或NADP⁺)反映进入呼吸链的电子的去向⁵、⁷。当ATP合成需求高时,绝大多数电子在细胞色素c氧化酶(复合物IV)催化的酶促反应中到达O₂;只有少数电子从上游移动载体辅酶Q(CoQ)过早泄漏,产生超氧化物和其他活性氧物种1⁷、3⁶(ROS)(图2a)。在由于供应增加(高NADH与NAD⁺比率)或需求减少(大质子动力势(∆p)和高ATP与ADP比率)而导致CoQ库过饱和的条件下,这些O₂的非酶促单电子还原概率急剧增加1⁷、1⁸、3⁶(图2a)。在清醒期间⁷,当热量摄入高但神经元的电活动减少,使其ATP储备充足时,dFBNs的线粒体易于以这种方式运作。事实上,使用基因编码的ATP传感器iATPSnFR和ATeam进行的测量显示,经过一夜睡眠剥夺后,dFBNs(而非投射神经元)中的ATP浓度比休息状态时高约1.2倍(图2b、c和扩展数据图4a、b)。当dFBNs受到觉醒性热刺激(通过向其树突释放多巴胺2、1⁴)抑制时,ATP浓度急剧上升(图2a、d);而当dFBNs自身受到刺激(模拟睡眠2)时,ATP浓度降至基线以下(图2a、e)。
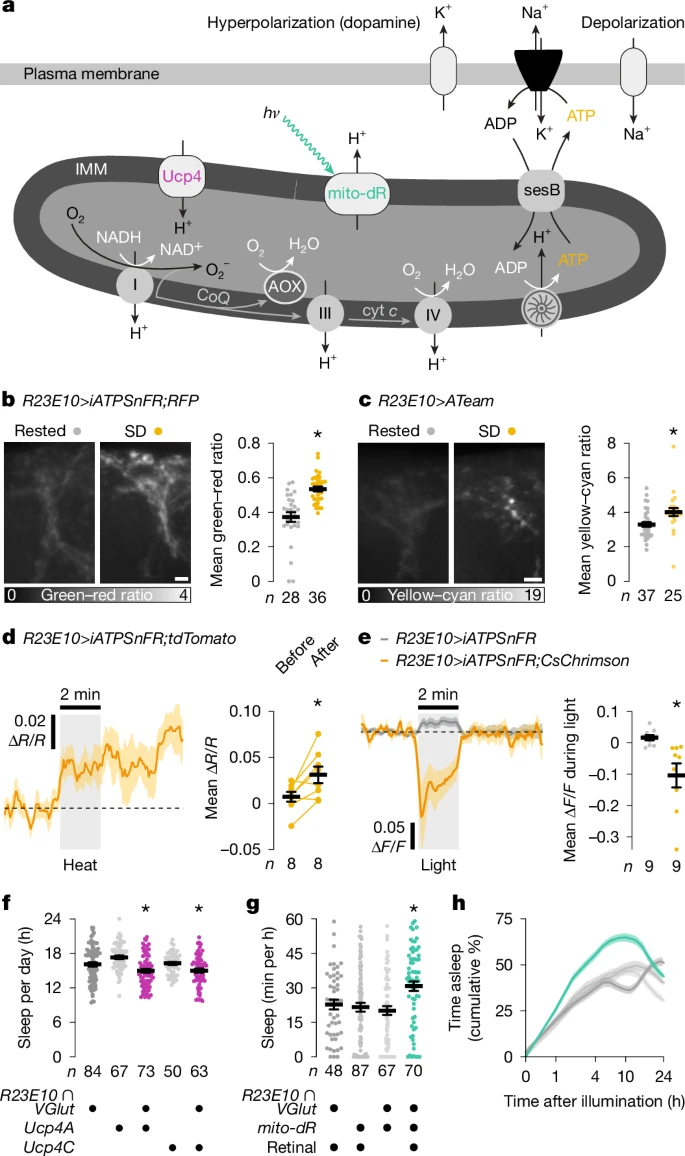
图2:线粒体电子过剩诱导睡眠
a. 质子泵复合物I、III和IV通过中间产物辅酶Q(CoQ)和细胞色素c(cyt c),将电子从NADH转移到O₂的能量转化为跨线粒体内膜(IMM)的质子电化学梯度∆p。Ucp4会释放线粒体内膜的质子梯度,而mito-dR在光照(hν)下会增强线粒体内膜的质子梯度。被泵出的质子返回基质时,会驱动ATP合酶的叶片旋转并产生ATP,ATP通过sesB离开基质,以交换细胞质中的ADP。神经元的ATP消耗具有活动依赖性,部分原因是质膜上的Na⁺-K⁺ ATP酶必须恢复因动作电位和兴奋性突触电流而消散的离子梯度。向CoQ供应的电子(相对于ATP需求)过剩,会增加复合物I和III处O₂被单电子还原为O₂⁻的风险。AOX可降低这种风险。b、c. 表达iATPSnFR加RFP(b)或ATeam(c)的dFBN树突在休息状态和睡眠剥夺(SD)果蝇中的总强度投影。发射比根据下方的图例进行强度编码,且在睡眠剥夺后增加(b中P < 0.0001,c中P = 0.0003;双侧Mann-Whitney检验)。d. 觉醒性热刺激提高表达iATPSnFR加tdTomato的dFBN中的ATP水平。在刺激前后立即的20秒窗口内对平均荧光进行量化(P = 0.0152,双侧配对t检验),并以共表达的tdTomato相对于刺激前基线的荧光强度比变化(∆R/R)绘图。e. 光遗传学刺激会减少表达iATPSnFR和CsChrimson的dFBN中的ATP,而在缺乏CsChrimson的dFBN中则不会(P = 0.0076,双侧t检验)。∆F/F是相对于刺激前基线的荧光强度变化。f. 表达R23E10∩VGlut-GAL4驱动的Ucp4A或Ucp4C的果蝇与亲代对照组的睡眠情况(方差分析(ANOVA)后进行Holm-Šídák检验,P ≤ 0.0381)。g、h. 光照后前60分钟的睡眠情况(g;Kruskal-Wallis方差分析后进行Dunn检验,P ≤ 0.0432)以及表达R23E10∩VGlut-GAL4驱动的mito-dR(有或无视网膜)的果蝇与亲代对照组的累积睡眠百分比(h;∆p光生成效应:P < 0.0001,时间×∆p光生成交互作用:P < 0.0001,混合效应模型)。星号表示与两个亲代对照组相比或在计划的成对比较中存在显著差异(P < 0.05)。数据为平均值±标准误;n为树突区域数量(b、c)或果蝇数量(d-h)。比例尺,5μm(b、c)。统计详情见补充表1。
然而,即使单个电子从CoQ库泄漏的概率很低,但代谢高度活跃的细胞(如神经元)由于通过其呼吸链的电子数量庞大,仍会产生大量活性氧(ROS)1⁷、1⁸、3⁶、3⁷。在这些细胞中,能量可用性(在清醒时达到峰值)与能量消耗(在睡眠活跃的神经元中于睡眠时达到峰值)之间的反循环关系,可能使dFBNs比许多处于清醒状态的神经元更容易出现更严重的电子泄漏,从而使其成为一个有效的预警系统,以防范广泛的损伤。由于多不饱和膜脂质尤其容易受到损伤⁵,dFBNs通过计算Hyperkinetic活性位点处脂质过氧化衍生羰基的还原量,间接估算线粒体电子泄漏的程度⁵。
多项证据表明,进入线粒体运输链的电子数量与为ATP生产提供燃料所需的电子数量之间的不匹配是睡眠的根本原因之一。首先,为CoQ库中的过剩电子开辟一条出口通道(通过为dFBNs的线粒体配备玻璃海鞘的替代氧化酶(AOX)⁶,该酶通过O₂的受控四电子还原产生水),不仅缓解了基础睡眠压力⁷,还纠正了那些清除过氧化脂质分解产物能力受损的果蝇的过度睡眠需求⁵。其次,增加dFBNs对电子的需求(通过过表达解偶联蛋白Ucp4A或Ucp4C,它们会使线粒体内膜(IMM)上的质子电化学梯度短路1⁵)(图2a),减少了睡眠(图2f和扩展数据图4c)。第三,用光子而非电子为ATP合成供能(通过照射线粒体靶向版本的光驱动古菌质子泵delta-视紫红质1⁶)(图2a和扩展数据图4d),使得dFBNs中NADH衍生的电子变得多余,并促使睡眠发生(图2g、h和扩展数据图4e、f)。
睡眠改变线粒体动态
鉴于这些丰富的证据,线粒体成为睡眠剥夺后dFBN基因表达重组的两个核心之一(图1c、d和扩展数据图2)也就不足为奇了。然而,编码线粒体蛋白的转录本上调是表明线粒体质量净增加,还是对细胞器损伤的代偿性反应,仍不明确。为了区分这些情况,我们用定位于基质的GFP(mito-GFP)标记dFBNs的线粒体,并在独立的一系列实验中,通过共聚焦激光扫描显微镜(CLSM)或光学光子重分配显微镜3⁸(OPRM)对神经元的树突区域进行成像。这两种光学显微镜都通过超微结构进行了验证:对去卷积图像堆栈的自动形态测量表明,尽管两种光学方法在某种程度上都高估了真实细胞器的大小,但dFBNs和触角叶单球嗅觉投射神经元中线粒体的大小比率与体积电子显微镜3⁹确定的比率相匹配(扩展数据图5a、b)。因此,尽管衍射无疑夸大了线粒体的绝对尺寸(尤其是在CLSM图像中),但仍可对相对形态差异做出有效的推断。
一夜的睡眠缺失,无论由机械扰动还是人为提高觉醒性多巴胺1⁴引起,都会减小dFBN线粒体的大小、长度和分支(图3a、b和扩展数据图5c-f),并导致线粒体外膜的关键分裂动力蛋白——动力相关蛋白1(Drp1)⁸⁻1⁰从胞质溶胶重新定位到线粒体表面(图3c)。OPRM的横向超分辨率约为衍射极限的两倍3⁸,它检测到线粒体数量随之增加(图3b),这表明发生了细胞器分裂,而CLSM未检测到这一点(扩展数据图5f,但关于绝对线粒体计数的解释,请参见方法部分的注意事项)。尽管对线粒体子体的计数看似不完整,但这两种方法对睡眠缺失后的线粒体分裂描绘了一致的图景(图3a、b和扩展数据图5c-f)。相比之下,触角叶投射神经元的线粒体没有留下睡眠历史的痕迹(扩展数据图6)。
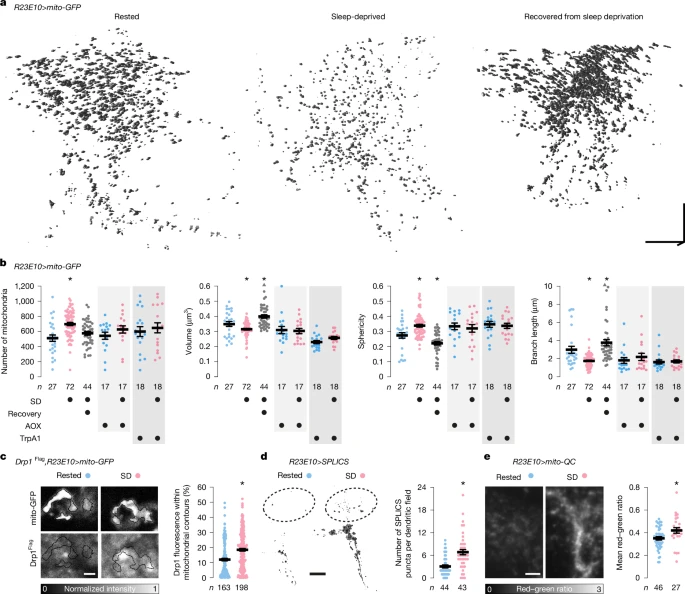
图3:睡眠历史改变线粒体动态
a、b. 休息状态果蝇、睡眠剥夺果蝇以及睡眠剥夺后允许恢复24小时的果蝇的dFBN树突在OPRM图像堆栈中自动检测到的线粒体的体积渲染图(a)和形态参数(b)。线粒体数量(方差分析后进行Holm-Šídák检验,P < 0.0001)、体积(Kruskal-Wallis方差分析后进行Dunn检验,P = 0.0470)、球形度(Kruskal-Wallis方差分析后进行Dunn检验,P = 0.0124)和分支长度(Kruskal-Wallis方差分析后进行Dunn检验,P = 0.0033)的睡眠历史依赖性变化被AOX的共表达所掩盖(双侧t检验或Mann-Whitney检验,P ≥ 0.2257),或被TrpA1的同时激活所掩盖(双侧t检验或Mann-Whitney检验,P ≥ 0.0625),并在恢复性睡眠后(过度)纠正(线粒体数量:P = 0.1551,所有其他参数:Kruskal-Wallis方差分析后进行Dunn检验,P ≤ 0.0302)。两个超过y轴限制的数据点以三角形绘制在图表顶部;平均值和标准误基于实际值。c. Drp1募集。表达R23E10-GAL4驱动的mito-GFP(上)和内源基因座的Drp1Flag(下)的果蝇的dFBN胞体的共聚焦图像平面。睡眠剥夺增加了自动检测到的线粒体轮廓内细胞抗Flag荧光(根据下方图例进行强度编码)的百分比(双侧Mann-Whitney检验,P < 0.0001)。d. 线粒体-内质网接触。通过对阈值化和去斑点共聚焦图像堆栈进行三线性插值获得的dFBN远端树突分支(虚线轮廓)中SPLICS斑点的等值面渲染(体素值128)。睡眠剥夺增加了每个树突区域的SPLICS斑点数量(双侧Mann-Whitney检验,P < 0.0001)。e. 线粒体自噬。表达mito-QC的dFBN树突的总强度投影。发射比根据下方的图例进行强度编码,且在睡眠剥夺后增加(双侧t检验,P = 0.0101)。数据为平均值±标准误;n为树突区域数量(b、d、e)或胞体数量(c);星号表示计划的成对比较中存在显著差异(P < 0.05)。比例尺,10μm(a)、2μm(c)、10μm(d)、5μm(e)。统计详情见补充表1。
AOX保护dFBN线粒体免受睡眠缺失诱导的碎片化(图3b和扩展数据图5e-g),这强调了清醒期间活性氧的产生⁷是触发分裂3⁶、⁴⁰的初始火花。同样,在机械性睡眠剥夺期间dFBNs的去极化(这会增加Na⁺-K⁺泵的ATP消耗,从而减少电子转向活性氧(图2a)),保护了其线粒体的形态(图3b和扩展数据图5e-g)。
线粒体分裂是线粒体在线粒体-内质网接触位点增殖⁴1和/或通过线粒体自噬脱落和清除功能失调的细胞器碎片⁴2的前奏(图3c)。睡眠剥夺刺激了这两个过程:锚定在线粒体外膜和内质网膜上的GFP片段的荧光重组(SPLICSshort,其本身对睡眠没有影响(扩展数据图5h))显示,睡眠剥夺果蝇的dFBNs中接触位点数量更多(图3d),而检测线粒体进入酸性自噬溶酶体的比率传感器mito-QC报告线粒体自噬增强(图3e)。线粒体-内质网接触点集中了分裂和融合机制⁴1、⁴3,它们的动态平衡决定了线粒体网络的稳态形态,并通过允许磷脂从内质网通过3、⁴来支持线粒体的生物发生。因此,睡眠剥夺的dFBNs中线粒体-内质网接触的丰富性(图3d)不仅可能反映了最近的线粒体分裂浪潮(图3a、b和扩展数据图5c-f),而且正如我们推测的编码线粒体蛋白的转录本的丰富性(图1c)那样,可能预示着在随后的恢复性睡眠期间线粒体的增殖和融合,这导致它们的体积、形状和分支长度反弹至基线以上(图3a、b和扩展数据图5e、f)。
线粒体动态变化改变睡眠若线粒体分裂与融合之间的平衡变化是纠正NADH供应与ATP需求不匹配(这种不匹配会导致睡眠压力升降)的反馈机制的一部分3⁶、⁴⁴、⁴⁵,那么在dFBNs中通过实验诱导这些稳态反应,应该会改变睡眠设定点:线粒体碎片化预计会减少睡眠时长和深度,而线粒体融合预计会增加睡眠时长和深度。为验证这些预测,我们对在线粒体动态变化中起核心调控作用的三种GTP酶进行了实验控制(图4a和扩展数据图7a-c):分裂动力蛋白Drp1(参考文献8、9、10),以及线粒体内膜和外膜的整合蛋白,分别称为视神经萎缩蛋白1(Opa1)和线粒体融合蛋白(或线粒体组装调节因子,Marf),它们在顺式和反式中的寡聚化可使相应膜融合⁹、11、12、13。与我们对线粒体质子动力势的操作(图2f-h和扩展数据图4c-f)一样,在这些实验中,R23E10-GAL4和交叉驱动子R23E10∩VGlut-GAL4(靶向中枢脑区真正的谷氨酸能dFBNs2(图1a))可互换使用(图4和扩展数据图8)。
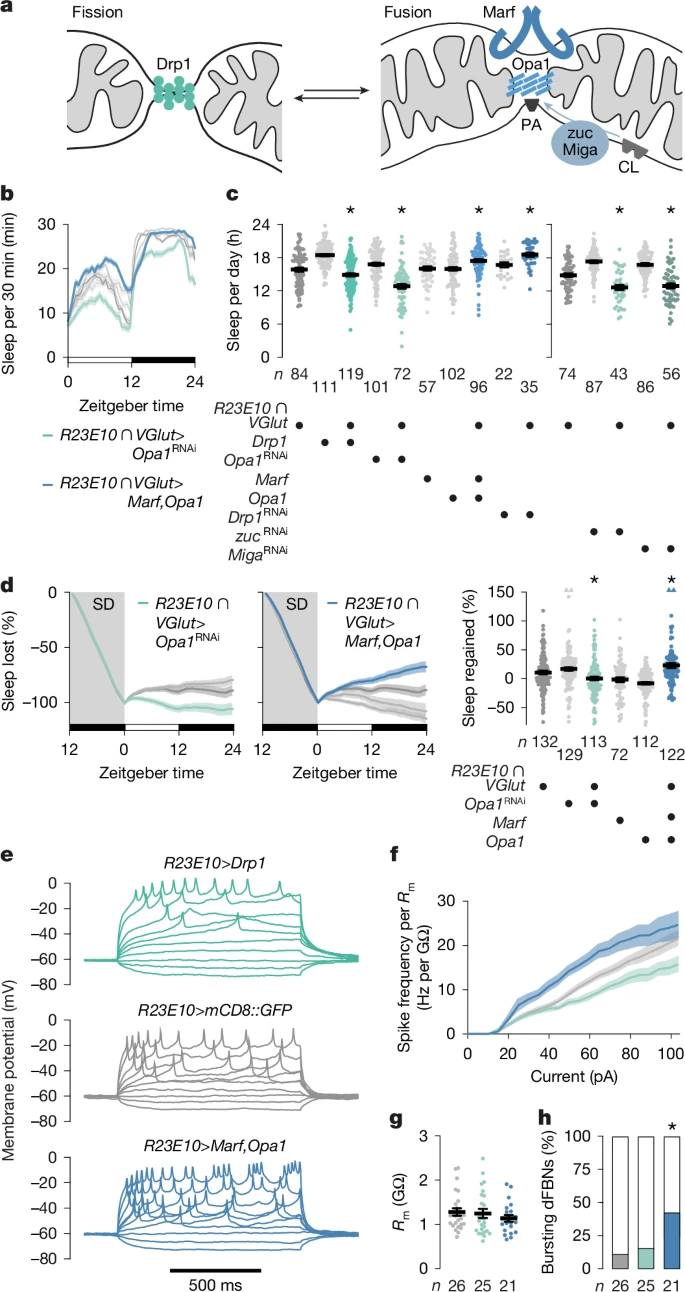
图4:线粒体动态变化改变睡眠
a. 线粒体分裂(绿色)和融合机制(蓝色)包括Drp1、外膜和内膜蛋白Marf与Opa1,以及线粒体磷脂酶D(mitoPLD)zuc,后者从心磷脂(CL)中释放磷脂酸(PA)。Miga刺激zuc活性和/或从其他膜提供磷脂酸。b、c. 表达R23E10∩VGlut-GAL4驱动的分裂或融合蛋白、靶向编码这些蛋白的转录本的RNAi转基因(左)或调控磷脂酸水平的转基因(右)的果蝇及其亲代对照组的睡眠曲线(b,基因型效应:P < 0.0001,时间×基因型交互作用:P < 0.0001,双向重复测量方差分析)和每日睡眠量(c)。增加分裂(绿色)或融合(蓝色)的操作以相反方向改变睡眠(GTP酶:方差分析后Holm-Šídák检验,P ≤ 0.0332;磷脂酸调控因子:Kruskal-Wallis方差分析后所有计划成对比较,P ≤ 0.0198)。d. 增加分裂(R23E10∩VGlut-GAL4 > Opa1RNAi,绿色)或融合(R23E10∩VGlut-GAL4 > Marf、Opa1,蓝色)的操作以相反方向改变睡眠反弹的时间进程(左图,基因型效应:P ≤ 0.0003,时间×基因型交互作用:P < 0.0001,双向重复测量方差分析)和剥夺后的睡眠反弹百分比(右图,基因型效应:Kruskal-Wallis方差分析后所有计划成对比较,P ≤ 0.0450)。四个超过y轴限制的数据点以三角形绘制在右图顶部;平均值和标准误基于实际值。e. 表达mCD8::GFP(灰色)和Drp1(绿色)或Marf加Opa1(蓝色)的dFBNs对电流阶跃的电压响应示例。f. 增加分裂(绿色)或融合(蓝色)的操作以相反方向改变膜电阻(Rm)标准化的尖峰频率(基因型效应:P < 0.0001,时间×基因型交互作用:P < 0.0001,混合效应模型,样本量见图g)。g. 膜电阻(基因型效应:P = 0.4806,Kruskal-Wallis方差分析)。h. Marf加Opa1的过表达增加产生动作电位爆发的dFBNs百分比(P = 0.0241,χ2检验;标准化残差+2.01)。数据为平均值±标准误;n为果蝇数量(b-d)或细胞数量(f、g)。星号表示计划成对比较中的显著差异(P < 0.05)。统计详情见补充表1。
通过过表达Drp1或RNAi介导的Opa1耗竭(以及在较小程度上对Marf的耗竭)使dFBN线粒体碎片化,会减少睡眠(图4b、c和扩展数据图8a-e),消除对睡眠剥夺的稳态反应(图4d和扩展数据图8f),并降低dFBNs中的ATP浓度,无论睡眠历史如何(扩展数据图7d)。使平衡倾向于线粒体融合则产生相反效果:dFBNs中Drp1的限制性敲低,或Opa1加Marf的过表达(或单独Opa1而非单独Marf的过表达),会增加基线睡眠和反弹睡眠(图4b-d和扩展数据图8a、f、g),并提高觉醒阈值(扩展数据图9a、b),且不会导致过表达伪影或明显的发育缺陷(扩展数据图9c、d)。当这些干预靶向投射神经元或肯扬细胞时,均未改变睡眠(扩展数据图10)。全神经元或神经胶质中Drp1或Marf的RNAi敲低后报告的无差别睡眠减少2⁴——在dFBNs中,这会双向改变睡眠(图4b-d和扩展数据图8a、f),反映它们在塑造线粒体方面的拮抗作用——若不平行测量线粒体(可能还有过氧化物酶体)的形态和功能,则难以解释。从表面上看,这可能暗示许多非dFBN细胞的融合-分裂循环与睡眠-觉醒周期无关,而是持续运作以混合和重新分隔线粒体内容物,用于维持或代谢控制⁴⁴、⁴⁵。
在dFBNs中,促进线粒体分裂或融合所产生的显著且相反的行为后果(图4b-d),与低或高睡眠压力的既定生物物理特征密切相关⁵、31。过表达Drp1的短睡眠者的dFBNs,其电流-尖峰频率函数比对照动物的神经元更平缓,而在嗜睡的Opa1和Marf过表达者中则相反(图4e-g),其dFBNs产生更多的促睡眠爆发2,作为其增强反应的一部分(图4e、h)。
睡眠剥夺大脑的一个显著特征是磷脂酸的耗尽⁵,磷脂酸是一种促融合的甘油磷脂⁴⁶。线粒体磷脂酸是心磷脂的裂解产物,由局部磷脂酶D(mitoPLD)生成⁴⁶(图4a)。为强调磷脂酸对融合反应的重要性以及线粒体融合对睡眠调节的重要性,R23E10∩VGlut或R23E10-GAL4限制性干扰mitoPLD西葫芦(zucchini)或外膜蛋白线粒体守卫蛋白(Miga)的表达(Miga稳定催化活性的mitoPLD⁴⁷和/或将磷脂(包括磷脂酸)从其他细胞膜转移到线粒体⁴⁸、⁴⁹),重现了当这些神经元的基于蛋白质的融合机制被RNAi靶向或被Drp1过表达拮抗时观察到的睡眠减少(图4c和扩展数据图8b)。
讨论
有氧代谢是一项革新,在24亿年前和7.5-5.7亿年前大气中氧气水平两次大幅上升中的第一次之后,真核生物得以最大化电子传递的自由能产出,为第二次氧气革命后寒武纪多细胞生命大爆发奠定了基础⁵⁰。高耗能的神经系统随之出现⁵1——显然,睡眠需求也随之产生⁵2。尽管睡眠此后可能获得了额外功能,如突触稳态或记忆巩固21,但将每日睡眠量与质量特异性氧气消耗相关联的经验幂律⁵3表明,睡眠在哺乳动物中也具有古老的代谢功能。该幂律中的异速生长指数是Euclidean几何缩放预期值的倍数,而非——这表明集中式网络(如血管和呼吸系统)的资源分配是其原因⁵3、⁵⁵。由于终端分支密度更高,这些网络为小型动物的每个细胞分配更多氧气,使其代谢比大型哺乳动物“更旺盛”,而大型哺乳动物的细胞受到供应限制⁵⁵。其代价是寿命更短,且一生中更大比例的时间用于睡眠。即使在物种内部,个体(包括核基因组相同的个体,如我们的同基因果蝇)的睡眠需求差异也可能部分源于含有线粒体DNA编码亚基的呼吸链的电子流阻力差异1⁸。事实上,强烈的疲倦感(与肌肉疲劳无关)是人类线粒体疾病的常见症状⁵⁶。
若睡眠确实是为满足代谢需求而进化的,那么控制睡眠和能量平衡的神经元受相似机制调控就不足为奇了。在哺乳动物下丘脑中,表达刺鼠相关蛋白(AgRP)的促食神经元和表达阿黑皮素原的厌食神经元的线粒体经历分裂和融合的反相循环⁵⁷。这些循环与小鼠的能量平衡变化相关⁵⁷,正如dFBNs中的线粒体分裂和融合循环与果蝇的睡眠平衡变化相关一样。线粒体融合后,AgRP神经元的电输出增加,以促进体重增加和脂肪沉积⁵⁷,正如线粒体融合后dFBNs的电输出增加以促进睡眠一样。从AgRP神经元中删除线粒体融合蛋白会损害食物消耗⁵⁷,正如干扰dFBNs中的线粒体融合会损害睡眠诱导一样。这些相似性表明,睡眠压力和饥饿都源于线粒体,电子流过各自反馈控制器的呼吸链,就像沙漏中的沙子一样,决定着必须恢复平衡的时刻。
转载本文请联系原作者获取授权,同时请注明本文来自孙学军科学网博客。
链接地址:https://wap.sciencenet.cn/blog-41174-1494166.html?mobile=1
收藏