
电驱动的有机氢化反应,以水为氢源
对本文的兴趣点是氢化,本人对氢气的生物学效应机制一直着迷,但又一直没有获得满意的结果。氢气发挥生物学效应存在两种可能,一是直接参与生物化学反应,二是间接参与生物化学过程。现在看起来,直接参与生物化学反应的可能性比较大。间接过程见于纯惰性气体如氙气的生物学效应,主要在于通过深入到分子结构内部,干扰其他化合物进入分子的速度。例如氙气能干扰甘氨酸进入NMDA受体结合部位,成为这种效应的理想阻断剂。且这种阻断剂没有化学活性,是非常安全副作用小的理想受体阻断剂。当然这种作用可能难以产生专一性或特异性。直接参与生物化学过程则需要打破氢分子共价键,这非常符合氢气作为生物代谢分子的特征,因为氢气可以被细菌生物合成,也可以被细菌生物分解利用。无论合成和分解,都存在氢气共价键的产生和解离。而这种过程会伴随化学能的利用和释放。但是由于氢气的产生和利用在微生物领域都存在特异性的酶催化过程,而高等生物似乎已经丢失了这种特异性的催化酶。但是没有特异性催化酶不等于没有类似活性的其他非特异性催化酶。没有催化酶也不等于没有可以结合氢气的其他类似生物活性分子。这些分子产生效应可能存在氢气的合成和分解类似过程。这种过程和工业上的氢化过程可能由类似特点。本文介绍利用水电解产生氢原子实现氢化的过程,是否会和氢气在生物体系内的过程由类似性。值得我们思考。
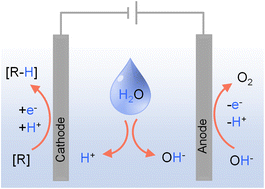
在有机合成中,氢化是一个关键过程,已经开发了多种催化策略来实现不同底物的有效氢化。尽管这些方法很有能力,但主要依赖于分子氢(H2)气体在高温和高压下的操作带来了挑战。其他替代氢源如无机氢化物和有机酸往往过于昂贵,限制了它们在大规模上的实际应用。相比之下,用水作为有机氢化的氢源呈现出一个有吸引力且可持续的替代方案,有望克服与传统氢源相关的缺点。结合电力作为唯一驱动力在环境条件下,使用水作为唯一氢源的氢化反应与环境可持续性目标相吻合,同时也提供了更安全、可能更具成本效益的解决方案。本文首先讨论传统氢源与水在氢化反应中的固有优势和局限性,然后介绍成功利用水作为氢源实现大量有机氢化转化的代表性电催化系统,重点介绍异质电催化剂。总之,过渡到使用水作为有机氢化的氢源代表了可持续化学的一个有希望的方向。特别是,通过探索和优化电催化氢化系统,化学工业可以减少对危险和昂贵氢源的依赖,为更安全、更绿色、能量密集度更低的氢化过程铺平道路。
1. 引言
氢化是现代化学合成的基石,在包括石油化工、煤化工、精细化工和环境工业在内的各个领域都具有重要意义。据估计,大约25%的所有化学过程至少包含一个氢化步骤。例如,加氢脱氮和加氢脱硫对于石油化工炼油厂中原油的净化至关重要,而选择性过程如不对称氢化为高效合成高价值精细化学品提供了有效途径。因此,寻求温和条件下的直接氢化方法已成为当代研究的重要焦点,这也符合提高化学工业整体可持续性和效率的需求。
虽然工业氢化主要依赖于使用H2作为主要氢源的热催化方法(图1A),但最近的研究焦点转变揭示了电催化作为一种可行的替代方案的潜力。这些新方法不同于传统的高能耗热催化过程,后者存在显著的可持续性问题。由于电力可以从光伏产生,将太阳能集成到电催化氢化中进一步增强了其环保和可持续性。
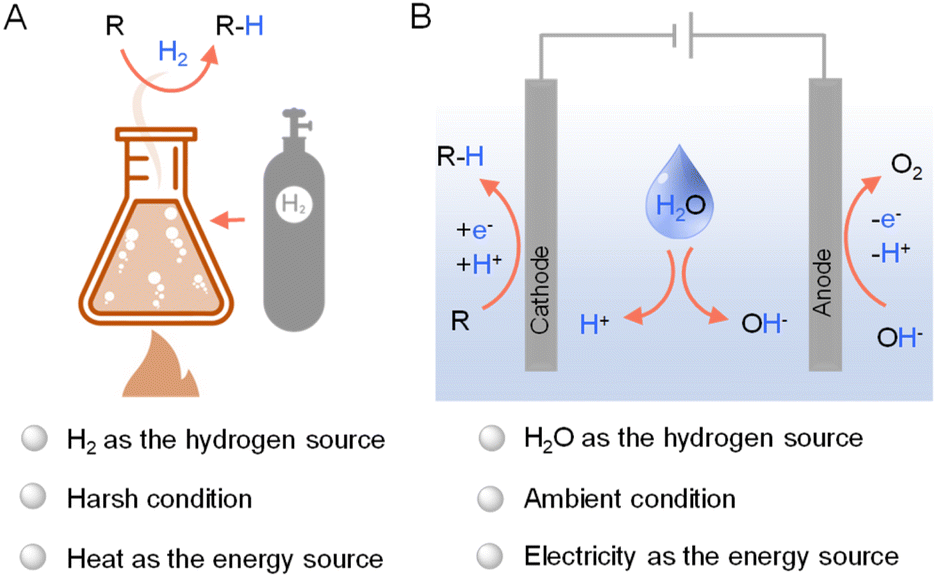
图1 (A) 使用H2的热催化氢化和使用水作为氢源的电催化氢化的一般表示。
除了能源输入,氢源在氢化反应中同样至关重要。尽管H2和其他分子氢供体(例如,甲酸、醇、胺、氢化物等)已广泛用于氢化反应,但它们自身的限制/约束促使人们寻求更绿色和更经济的替代方案。可以说,最绿色且成本最低的氢源是水。实际上,结合电催化技术,水已被成功用作多种有机氢化应用中的多功能氢源。本文旨在深入探讨以水为主要氢源进行氢化反应的原理,这是使用异质电催化剂进行绿色氢化领域的一个有希望的方向。强调水相对于传统氢源的独特优势,我们将对不同的氢化策略进行比较分析,然后介绍使用水作为氢源的各种氢化反应的代表电催化系统。通过研究这些新兴方法,我们旨在突出基于水的氢化作为一种可持续且高效方法的潜力,适用于化学工业。
2. 不同氢源用于有机氢化的比较
在氢化过程中,除催化剂外,所需的基本元素是氢源。以下部分将简要讨论包括H2在内的常用氢源的优势和局限性。
2.1 分子H2
H2在工业热催化氢化过程中占据主导地位,提供几个独特的优势。H2的高反应性是一个主要好处,使各种官能团的有效氢化成为可能。这种反应性允许烯烃、炔烃和羰基化合物等多种物质的氢化。另一个优点是使用H2的反应简单且清洁,与其他还原剂相比通常产生较少的副产品。这方面在产品纯度和反应后纯化方面特别有利。此外,H2可以与各种异质和均质催化剂配对,使整个过程在经济和环境上更具有吸引力。此外,H2在不对称氢化中的应用为手性分子提供了途径,这在药物合成中至关重要。最后,使用H2符合绿色化学原则,特别是当其源自可再生资源(如生物质)时,有助于可持续的化学实践。
尽管有这些优势,H2在氢化反应中也存在某些局限性。因为氢化需要氢气原子或氢化物,所以H-H键的均裂或异裂是涉及H2的氢化反应的先决条件,这可能需要大量能量。此外,与其使用相关的安全顾虑是一个主要挑战。H2高度易燃且易爆,尤其是在高压下,需要严格的安全协议和专业设备。这可能导致操作成本和复杂性的增加。此外,许多氢化过程需要高压,这可能限制反应的规模并需要昂贵的反应器。另一个缺点是选择性问题。尽管H2对广泛的氢化有效,但其非特异性可能导致过度氢化或多个官能团的还原,这在复杂分子合成中是不可取的。目前市场上主要从化石燃料中提取的H2的依赖也引发了环境担忧。事实上,由于工业H2主要是通过蒸汽甲烷重整生产的,这可能包含一定量的CO杂质,催化剂中毒并不是一个可以忽视的问题。
2.2 无机氢化物
氢化反应也可以使用许多无机氢化物进行,特别是在相对小规模的应用中。无机氢化物,如LiAlH4和NaBH4,在实验室研究中广泛用于有机氢化反应,每种都有独特的优点和局限性。LiAlH4在相对较温和的条件下非常有效地还原各种官能团,如羰基和羧酸。然而,其极高的反应性和对水分的敏感性需要无水条件和小心处理。NaBH4的反应性低于LiAlH4,提供了更安全的处理和与一些质子溶剂的兼容性,但其效力较低,主要还原醛和酮。使用这些无机氢化物通常需要化学计量量,导致废物处理问题,尤其是铝盐。它们的非选择性也可能是一个问题,在更需控制还原的复杂合成中。每种氢化物都提供了一种反应性和选择性的平衡,针对特定应用最大化其益处同时最小化缺点,以优化有机合成。
2.3 有机氢供体
除了H2和无机氢化物外,还有大量的有机分子,如醇、醛、甲酸、胺等,被用作各种有机氢化反应的氢源。图2A展示了用作氢化反应氢源的那些代表性有机分子,每个都有其自身的优点和局限性。
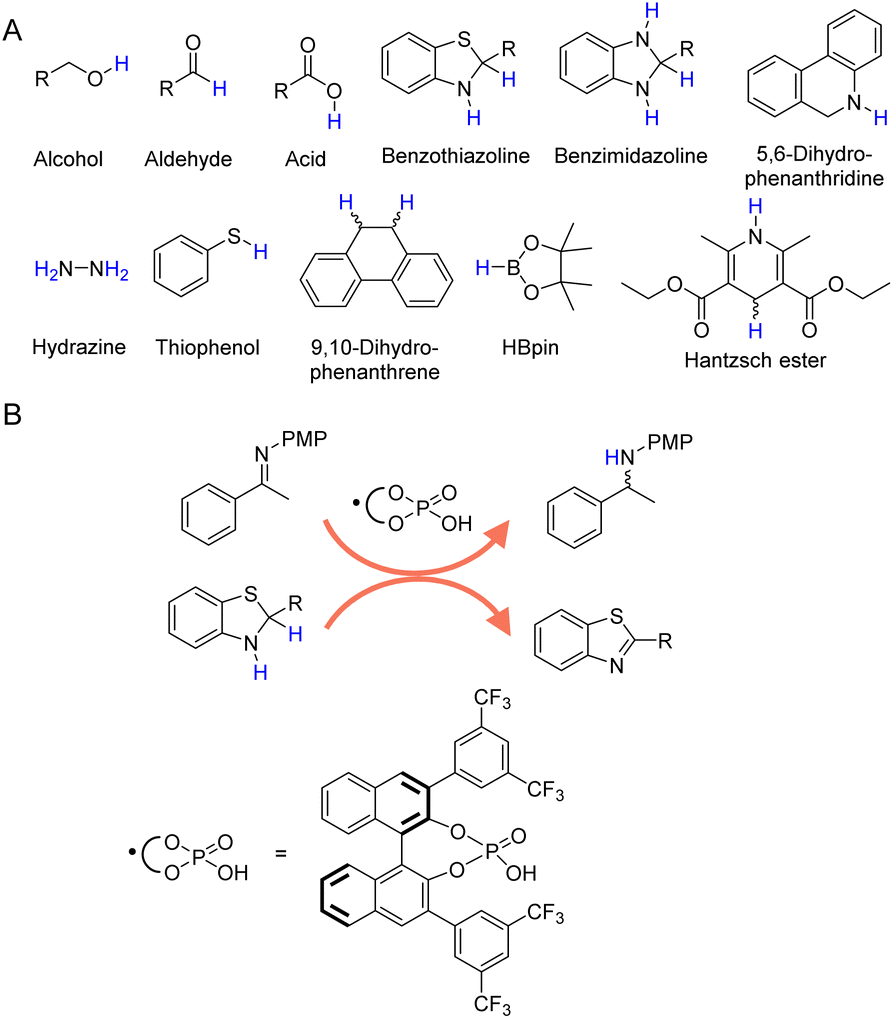
图2 (A) 有机氢化过程中使用的选定有机分子。(B) 苯并噻唑啉与手性磷酸结合显示出通过释放氢气形成更稳定的苯并噻唑的还原能力。
例如,在氢化过程中使用醇类呈现了作为氢供体和溶剂的独特组合,使得这些方面的效应交织在一起。各种醇类,如甲醇、43乙醇、38,40,44,45异丙醇、46–52 2-丁醇、38甘油和苄醇,53具有许多优点,包括广泛的可获得性、可负担性、潜在的可再生性质、方便的运输和储存。54使用醇类通常可以导致更具选择性的氢化过程,这归功于其特定的反应性。然而,也存在显著的缺点。与纯H2相比,这些醇类释放氢气的效率通常较低,可能导致更高的能量消耗和较低的反应速率。对于较大的醇类,如2-丁醇,由于它们更复杂的分子结构,这种低效率可能会加剧。55,56此外,使用醇类通常需要更复杂的催化剂系统,这可能增加反应的整体成本。还有副产物形成的挑战,可能使所需产品的纯化过程复杂化,并影响整体可持续性。57
同样地,甲醛和甲酸41,42,57–64也已被广泛用作氢化反应的氢源。作为一种容易获得且成本效益高的商品化学品,甲醛因其高氢含量而受到高度评价。然而,其毒性要求严格的安全协议和专业设备,增加了操作复杂性和成本。此外,甲醛的反应性可能导致副反应和选择性挑战,可能使所需产品的纯化过程复杂化。另一方面,甲酸的毒性明显较低,使其成为实验室和工业氢化使用的更安全替代方案。此外,甲酸与多种催化剂兼容,可用于不同类型的氢化反应,包括烯烃、炔烃和羰基的还原。然而,其低反应性通常需要大量或更严格的反应条件才能与其他氢源达到相当的结果。在某些情况下,甲酸可能参与副反应,潜在地导致不希望的副产品。57,65甲酸及其衍生物(如CO)导致的催化剂失活风险不容忽视。
在催化氢化的领域内,利用有机氢供体如苯并噻唑啉(图2B)、66,67苯并咪唑啉、68,69 5,6-二氢菲啶、70肼、71–73硫酚、74 9,10-二氢菲、75 HBpin以及汉斯酯(NAD(P)H的分子模拟物)77–81提供了一种优势与劣势微妙平衡的方法。这些化合物以其能够提供受控和选择性的氢转移而著称,类似于生物系统中的酶促过程。这种选择性对于那些需要在特定位置进行选择性氢化而不干扰其他敏感官能团的反应尤其有利。然而,高昂的成本和可获得性可能是限制其在大规模应用中的重要因素,特别是对于像汉斯酯和HBpin这样的化合物。虽然最近几份报告展示了一些这些分子氢供体的电催化再生,82但产生副产品的潜在风险是另一个关键问题,往往需要额外的纯化步骤,这增加了整体过程的复杂性和成本。因此,尽管这些有机氢供体的使用为催化氢化提供了一种创新方法,但必须权衡其益处与经济、操作和环境影响。
作为无碳化合物,氨(NH3)和铵盐也被广泛用于各种氢化反应中作为氢源。40,83 NH3是一种特别密集的氢源(约17.6 wt%),使其成为有效的储氢和运输方式。它也相对容易在温和条件下液化,这增强了其工业应用的实用性。此外,NH3和铵盐广泛可得且可大规模生产,通常成本低于纯H2。在此背景下,NH3已成功用作烯烃、炔烃和酮的电催化氢化的氢源。84–86例如,图3展示了在阴极使用NH3作为氢源的烯烃电催化氢化方案。84除了NH3,有机仲胺87和铵盐(如三乙铵甲酸盐88)也在各种氢化反应中被用作氢源。39,89然而,使用NH3和铵盐进行氢化也面临重大挑战。在热催化条件下,从这些化合物中释放氢气通常需要高温和高压,导致能源消耗增加和反应范围受限。NH3和铵盐的催化分解可能产生不希望的副产品,导致产品纯化困难和潜在的环境问题。鉴于它们的有毒和腐蚀性,通常需要专门的设备和安全协议。
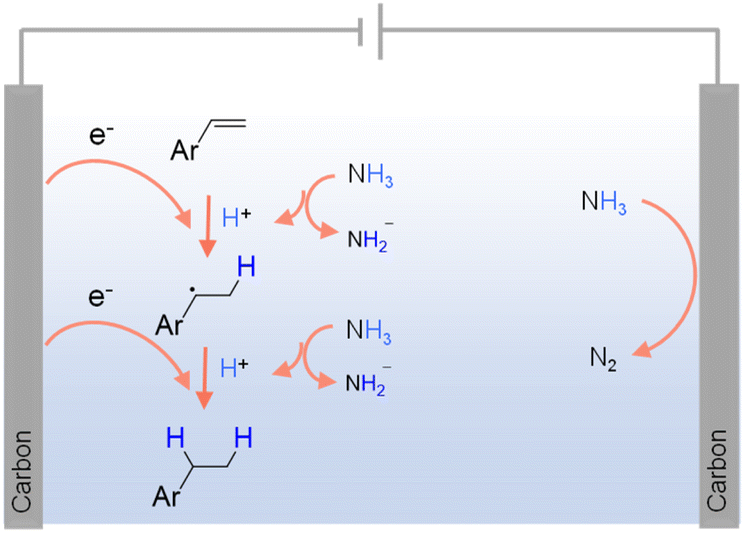
图3 使用NH3作为氢源的烯烃电化学氢化可能机制。83
2.4 不同的活性氢物种(H+、H˙和H−)
如前所述,有机氢化反应中可能涉及不同的氢物种。实际上,质子(H+)、氢原子(H˙)和氢负离子(H−)都是可能的氢物种,具体取决于特定的氢化机制。如图4所示,电力驱动的氢化反应主要遵循两条路径:(i)氢原子转移(HAT)和(ii)质子耦合电子转移(PCET)。遵循HAT机制时,首先在阴极表面形成吸附的氢原子,随后将添加到底物上,90–92类似于有机金属催化中的HAT步骤。34相反,遵循PCET机制时,H+和e−会同时转移到底物上。93–96具体的氢化途径将由许多因素指示,包括电催化剂、微环境、电位、pH等。
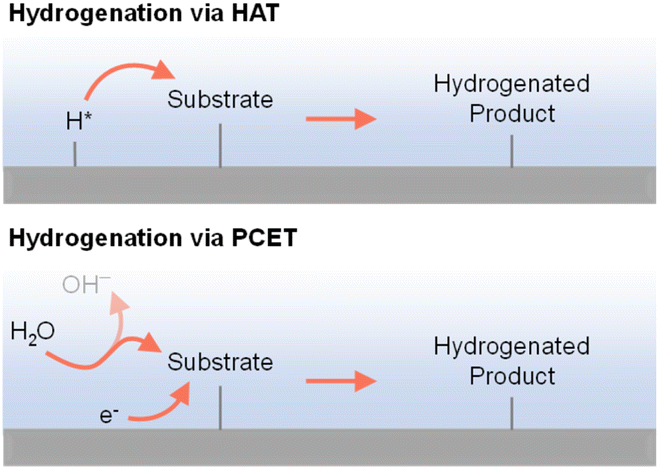
图4 通过氢原子转移(顶部)或质子耦合电子转移(底部)过程进行的电力驱动氢化。97
3. 使用水作为氢源的电催化氢化
鉴于前述H2和各种无机/有机氢源的局限性,人们越来越有兴趣探索更环保的替代方案,其中水作为一种环境和经济上可行的选择脱颖而出。事实上,使用水作为氢源的电催化氢化已经成为传统热催化氢化的一种典范替代方案,它提供了一种在环境条件下利用电力作为主要驱动力合成氢化产品的高效且可持续的途径。
对于容易直接还原的有机底物,它们的氢化反应可以在没有任何电催化剂的情况下发生。例如,Werz等人报告了通过电还原,使用水作为氢供体进行苄基烯烃的位点选择性氢化。98如图5所示,经过单电子转移(SET)过程,苄基烯烃在阴极被直接还原形成自由基阴离子,随后通过H2O的质子化作用淬灭。另一个SET步骤在阴极从自由基中间体中产生一个阴离子,该阴离子在质子化后生成所需产物。另一方面,阳极上水的氧化可以直接生成O2和H+。
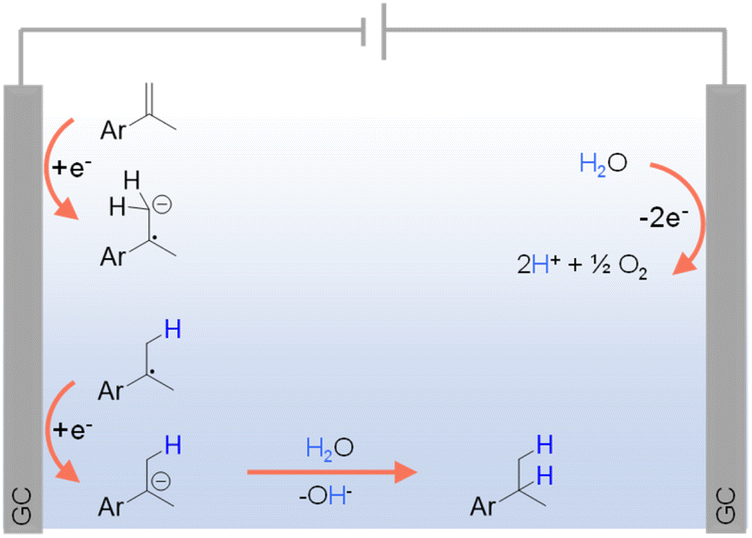
图5 使用水作为氢源对不同烯烃的氢化(GC = 玻璃碳),使用0.02 M TBAPF6在CH3CN/H2O(v/v = 4/1)中作为电解液(TBA = 四正丁基铵)。98
然而,大多数电化学氢化反应都需要电催化剂来促进这些过程。在大多数情况下,在氢化步骤之前,将水分解以产生某种活性形式的氢(例如,原子氢、质子、氢负离子和分子二氢)是至关重要的。为此,研究人员采用了多种电催化策略。接下来的段落将讨论影响电催化氢化性能的因素,并介绍几种能够减少能量输入同时使电催化氢化反应速率翻倍的有前景的策略。
3.1 电催化剂在氢化中的影响
许多已报道的电催化剂实际上由具有已知热催化氢化活性的金属组成,如Pd、Pt、Ru、Cu和Ni,所有这些金属都能在还原条件下形成活性氢物种。97,99–101由于观察到的电催化活性通常与电催化剂的活性表面积成线性比例,因此高度多孔的电催化剂一直备受青睐。因此,实现小颗粒尺寸的纳米结构是电催化氢化中的常见策略。极端情况是所谓的单原子电催化剂,它们不仅展示了最大化的电催化剂原子利用效率,特别是对于昂贵的贵金属,还可能表现出独特的活性。102–104
因为催化剂发挥的关键作用,理解电催化剂内在活性的来源对于开发有效的电催化氢化系统至关重要。一个典型的例子来自Koper小组,他们全面比较了Pt催化剂三种不同晶面对丙酮电催化氢化的活性。105在他们的研究中使用了单晶Pt电极。DFT计算表明,由于其表面Pt原子的高配位数,丙酮与Pt(111)和Pt(110)的相互作用在能量上是不有利的。相反,丙酮还原发生在Pt[(n−1)(111)×(110)]和Pt[(n+1)(100)×(110)]的台阶位点上,尽管对于异丙醇或丙烷的选择性有所不同。如图6所示,丙酮还原的两条途径通过形成共同的中间体*CH3COHCH3进行,但对其后续转化有所不同。换句话说,这两条途径在形成异丙醇前分叉。一旦形成异丙醇,进一步氢化为丙烷就不可能了。事实上,选择性源于*CH3COHCH3质子化(图6顶部路线)与其C–O键断裂(图6底部路线)的相对容易程度。研究发现,丙酮主要在Pt[(n−1)(111)×(110)]的台阶位点被还原为异丙醇,而在Pt[(n+1)(100)×(110)]的台阶位点则生成丙烷。这项研究的指导可以应用于其他高级脂肪族酮的电催化氢化。
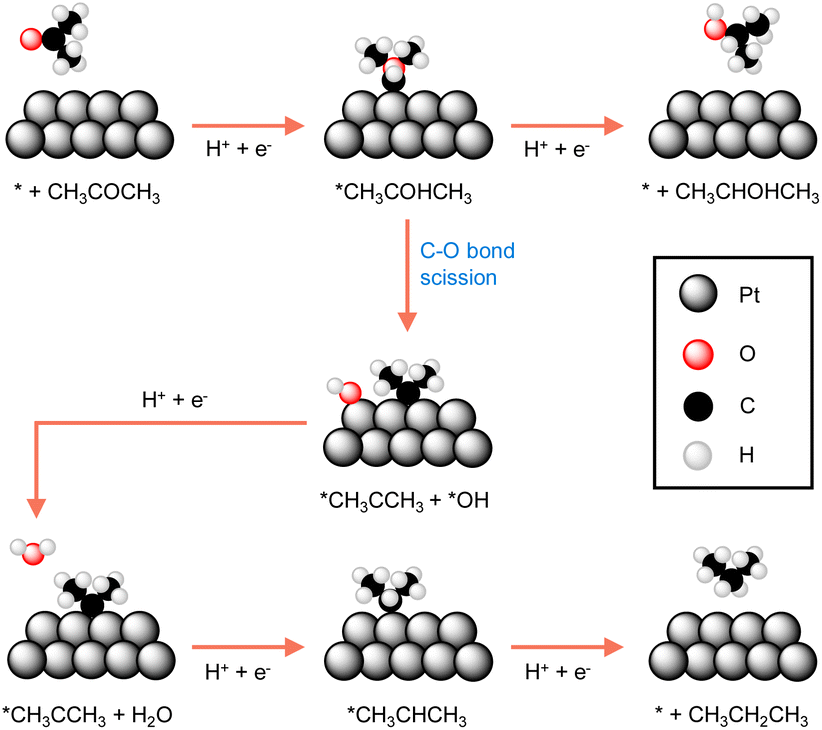
图6 计算得到的在Pt电极上丙酮还原为2-丙醇和丙烷的最有利反应途径。105
除了前述的贵金属如Pt,地球上丰富的第一过渡金属也在电催化氢化有机物中得到了广泛应用。例如,广泛用于电催化CO2和CO氢化的Cu基复合材料,也展示了对乙炔半氢化为乙烯(石化工业中最重要的烯烃)的优异性能。不幸的是,约1%的乙炔不可避免地作为杂质在用于乙烯生产的石脑油裂解器中生成,且共产生的乙炔杂质会严重毒化聚合催化剂,不利地降低目标聚合物的质量。与热催化乙炔半氢化相比,电催化半氢化因其环境友好性和经济效率而呈现出一种有前景的替代方案。理想的乙炔半氢化电催化剂应具备适当的乙炔吸附能但HER性能较差。同时,产生的乙烯应易于从电催化剂表面脱附,否则会发生过度氢化为乙烷。邓等人证明了Cu是一种高效的乙炔选择性半氢化电催化剂。通过原位X射线吸收精细结构实验研究了Cu催化剂在有无乙炔吸附时的电子性质。结果表明,乙炔在Cu表面的吸附导致催化剂氧化态增加,这是由于电子从Cu转移到吸附乙炔的反键π轨道所致。这一结果也通过原位拉曼光谱表征吸附的乙炔得到证实,其中观察到吸附在Cu上的C–C键减弱。DFT计算也被用来验证这些实验结果。考虑了包括(111)、(100)和(110)在内的三种Cu晶面,发现在所有晶面上的空洞位点都强烈吸附乙炔,其吸附自由能远高于氢吸附。基于巴德电荷分析,Cu的价电子显著减少,吸附的乙炔增加了多达0.65个电子,证实了电子从Cu表面向乙炔的转移。图7展示了吸附结构的差分电荷密度,显示了Cu吸附位点周围的电荷耗尽区域以及Cu表面上吸附乙炔周围的电荷积累区域。
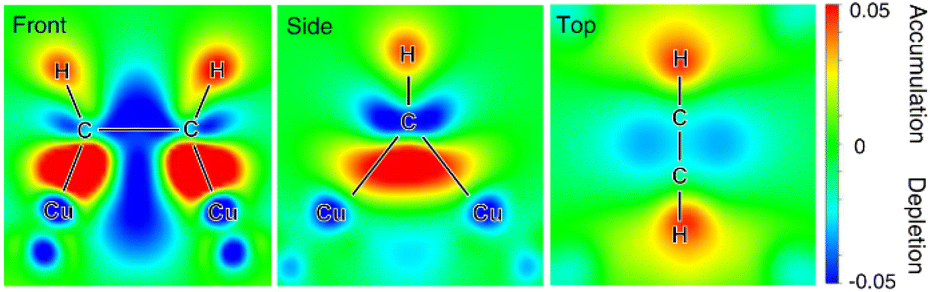
图7 乙炔吸附在Cu表面的电荷密度差异,前视图、侧视图和顶视图。红色和蓝色分别表示电荷积累区域和耗尽区域。该图改编自文献109,经Nature Publishing Group许可,版权所有2021。
其他通过改良制备方法的Cu基电催化系统实现了进一步的性能提升。例如,电沉积的Cu树枝状结构可以将乙烯流中的乙炔杂质水平从1×104 ppm降至4 ppm。张等人报道了一种在涂有气体扩散层的碳纸上合成的Cu纳米颗粒电催化剂,在工业相关电流密度为0.5 A cm−2时,实现了高C2H4产率70.15 mmol mg−1 h−1和法拉第效率97.7%,根据其技术经济分析,这在工业上是有利可图的。
对于更具挑战性的芳香族氢化反应,也有报道称通过各种掺杂剂改性的电催化剂可以实现。例如,一种掺氟的钴(Co–F)电催化剂能够利用水作为氢源促进喹啉(1a)的电催化氢化,生成1,2,3,4-四氢喹啉(2a)。图8A揭示了表面F的存在可以显著增强1a的吸附,这将促进1a与电催化剂之间的电子传递。此外,2a的吸附能远低于1a,这意味着产物更容易从Co阴极上脱附(图8B)。而且,Co–F上形成H*的吉布斯自由能比纯Co更负(-0.46 vs. -0.38 eV,图8C)。这表明Co–F阴极有利于通过水解离生成H*,这将有助于1a的氢化。
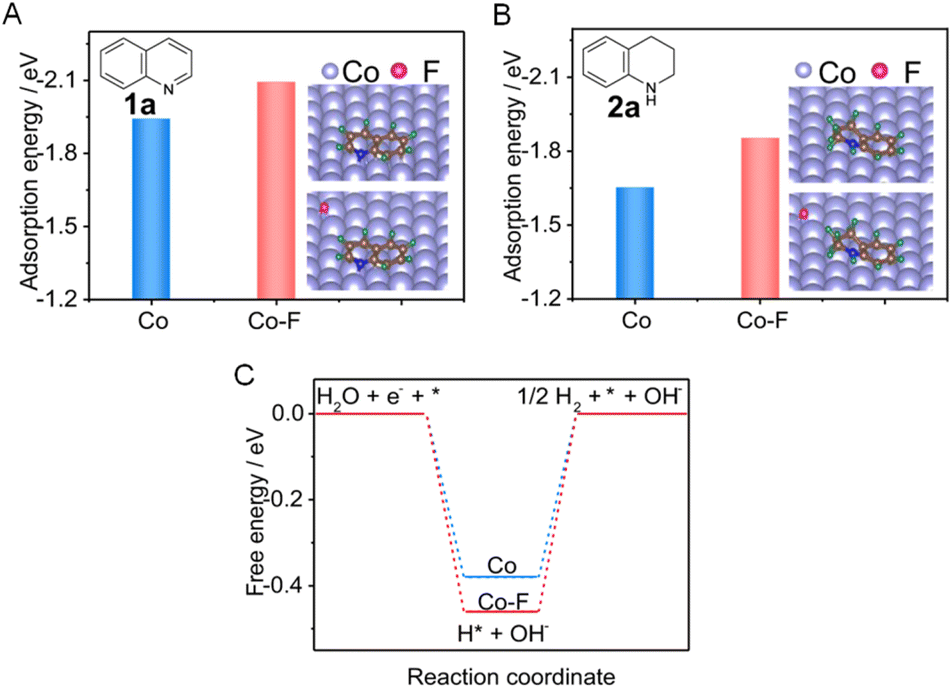
图8 (A和B) 分别比较了1a和2a在纯Co(A)和Co–F(B)上的吸附能(插图:1a和2a的稳定吸附构型)。(C) Co与CoF上H*形成的计算自由能。该图改编自文献112,经Nature Publishing Group许可,版权所有2022。
一种双金属PtRu电催化剂被报道用于在酸性电解液中高效地将苯甲酸氢化为环己烷羧酸。许多苯甲酸衍生物的环结构可以以类似的方式氢化,尽管法拉第效率一般。实际上,大量主要由地球丰富元素组成的电催化系统已被探索用于各种有机底物,特别是生物质衍生中间体的电催化氢化,其中水是主要的氢源。
3.2 微环境对氢化的影响
除了明智选择高效的电催化剂外,精细控制电催化剂活性位点周围的微环境同样重要,它决定了氢化效率和选择性。微环境可以通过改变pH、溶剂、支持电解质、添加剂等进行调节。
由于电催化氢化需要通过质子或水还原在电极表面生成吸附氢,因此存在一个可能的竞争反应,即有机反应物的直接还原。在后一种情况下,电子在反应物和阴极之间传递,随后在溶液中发生质子化,可能导致副产物的产生。为了尽量减少有机底物的直接电还原,控制有机反应物、质子和电极之间的界面相互作用至关重要。李等人以铜电极上糠醛的电催化氢化为模型反应,进行了彻底的研究以区分电催化氢化和直接电还原的机制。使用带有自组装巯基单层(如3-巯基丙酸(MPA)和2-巯基苯并噻唑(MBT))的铜电极,以确定有机反应物和阴极表面之间的直接相互作用的必要性以及非均相电子转移的性质。发现在覆盖有MPA和MBT的电极上,2-糠醇和2-甲基呋喃的形成大大受到抑制(图9),这表明糠醛与铜电极之间的直接相互作用对于其电催化氢化和加氢裂解是必需的。然而,氢化糠醛的形成不受影响,表明电还原机制中的第一个电子转移是一个外球过程,对电极表面性质或催化活性不敏感。在电极上使用表面活性剂改性也被用来实现高选择性的电催化半氢化炔醇为烯醇,而水是氢源。
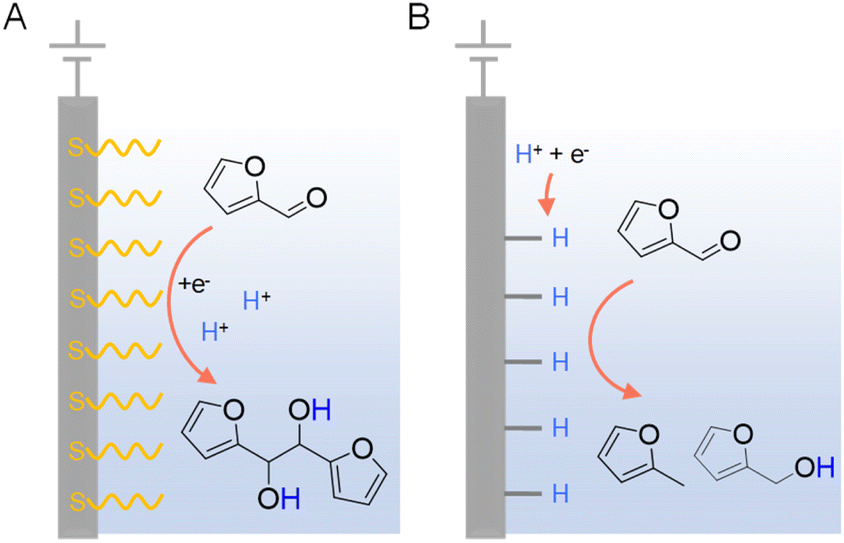
图9 在酸性电解液中铜电极上电催化糠醛还原的两种不同机制:(A)直接电还原和(B)电催化氢化。典型的电解液是0.5 M硫酸(pH = 0.5)或0.5 M硫酸盐溶液(pH = 1.4–3.0),含有25% v/v CH3CN。
除了有机底物与电极的直接相互作用外,有机底物在电极表面的特定吸附构型也会影响电催化氢化的结果。实际上,由于电催化氢化过程中需要质子源,局部H+浓度不仅在控制竞争性析氢反应(HER)方面起关键作用,也影响有机反应物的吸附构型。例如,乔等人通过将支持电解质H2SO4的浓度从0.1 M变化到1.0 M,观察到使用基于Pt的电催化剂对丙酮电催化氢化及氢解生成异丙醇和丙烷的产物分布有显著差异。为了阐明所观察到的结果,进行了详细的电化学研究、原位傅里叶变换红外光谱研究以及操作中的密度泛函理论计算(见图10)。得出的结论是,局部pH环境在很大程度上决定了丙酮的吸附构型,从而影响了丙酮转化为丙烷氢化(APH)的效率。当局部H+浓度低时(如0.1 M H2SO4),丙酮的吸附构型依赖于电势,当电势为正时,通过Pt@O键垂直吸附。在这个正电势下,APH的活性相当低,因为吸附的H*数量有限。一旦施加更负的电势,丙酮在Pt上的平面吸附构型更为有利,这不利于APH。相反,HER成为主导反应。然而,在高度酸性环境(1.0 M H2SO4)中,由于其较强的吸附强度,与平面构型相比,丙酮会采取垂直构型。因此,实现了更高的APH选择性。尽管在高度酸性环境中HER通常非常活跃,但竞争性的H2产生被严重抑制,因为在丙酮垂直吸附时,大多数吸附的H*参与APH而不是HER。可以预见,通过改变局部pH来改变反应物/中间体的吸附构型这一新策略,可适用于优化涉及多种物种的其他电催化反应的选择性。
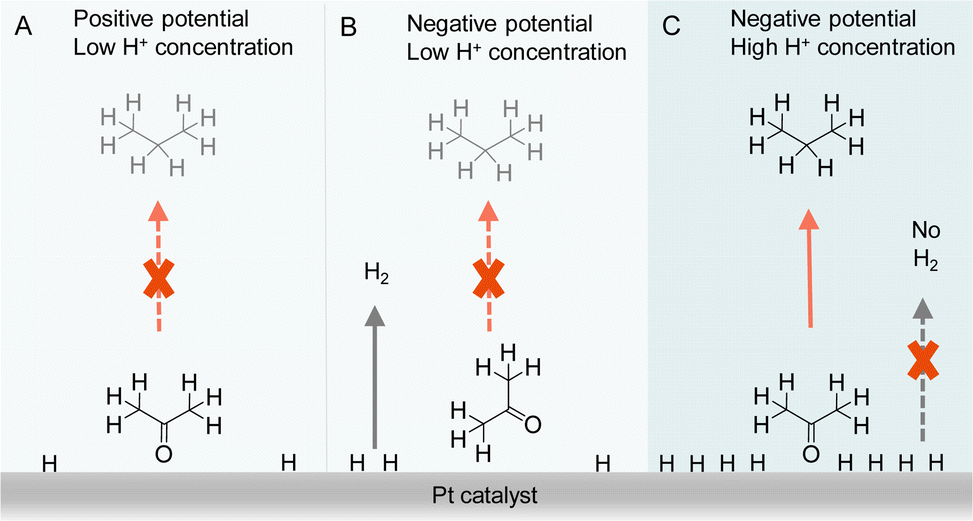
图10 在低H+浓度电解液(0.1 M H2SO4)中,正电势下丙酮转化为丙烷氢化(A),负电势下丙酮转化为丙烷氢化(B),以及高H+浓度电解液(1 M H2SO4)中,负电势下丙酮转化为丙烷氢化(C)。
3.3 配对电解在氢化反应中的优势
在水相中进行的传统电化学氢化反应中,阳极上常见的反反应是氧气析出反应(OER),这需要高电压输入,而只产生氧气这种低价值产物。因此,配对电解作为一种有前景的策略应运而生,它通过将目标电催化氢化反应与一个有价值的阳极反应相结合,从而替代OER。配对电解在实验室和工业规模上都是一个迅速发展的领域,这归功于其在两个电极同时进行有效反应时所带来的能量效率提升。特别是,氧化利用生物质衍生中间体化合物(例如5-羟甲基糠醛(HMF)、糠醛等)已被广泛用作阳极反反应的有效选项。
例如,孙等人受到Ni基复合材料上电催化OER中活性Ni3+中间体的启发,开发了NiBx作为在碱性水条件下将HMF氧化为2,5-呋喃二甲酸(FDCA)的有效电催化剂(见图11A)。他们进行了详细的电化学和光谱研究,以确认在NiBx上电化学生成的Ni3+(作为NiOOH)是氧化HMF到FDCA的活性物种。NiBx电催化剂也被用于阴极,用于对硝基苯酚的氢化为对氨基苯酚。因此,在整个配对电解过程中,使用水作为氧源和氢源分别获得了两种有价值产品(即FDCA和对氨基苯酚)。这样的配对电解池还与太阳能电池集成,作为一个独立的反应器用于阳光驱动的生产,具有高转化率和选择性(>90%)。
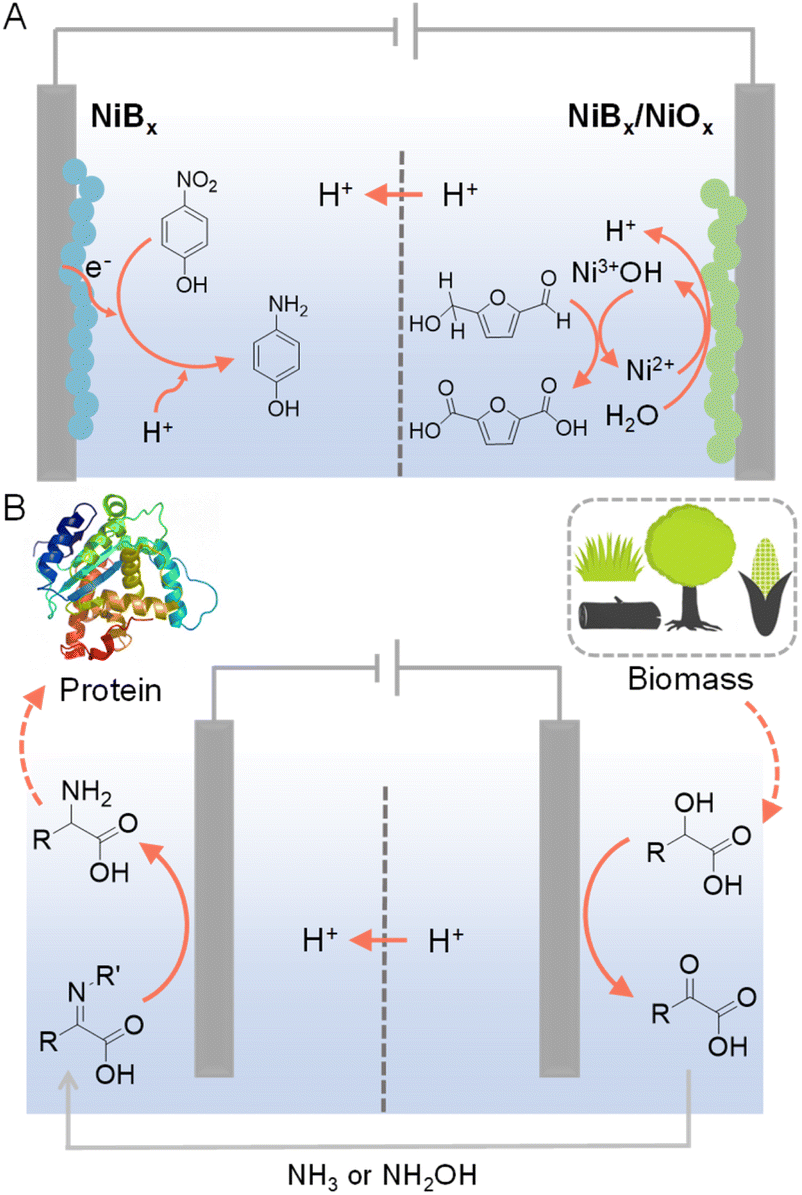
图11 (A)HMF氧化与对硝基苯酚氢化为对氨基苯酚的配对反应。134 (B)生物质衍生的α-羟基酸通过级联方式生成氨基酸。对于α-羟基酸的选择性氧化,使用N-羟基邻苯二甲酰亚胺(NHPI)或(2,2,6,6-四甲基哌啶-1-基)氧基(TEMPO)作为氧化还原介质,碳纸作为阳极,0.1 M LiClO4 CH3CN/H2O(v/v = 2/1)作为电解液。对于α-酮酸的电还原胺化反应,钛箔被用作阴极。阴极电解液是在室温下将20 mM α-酮酸与1.2–1.5当量的氨水(NH3 aq.)或盐酸羟胺(NH2OH·HCl)在0.1 M LiClO4 CH3CN/H2O(v/v = 2/1)中搅拌过夜形成的。135
与上述并行配对电解不同,后者利用两种反应物在两个电极上产生两种有价值产品,级联配对电解也可以采用以最小化电压输入同时生成高价值产品的方式,其中电催化氢化是两个连续步骤之一。例如,从生物质衍生的α-羟基酸开始,使用NHPI(N-羟基邻苯二甲酰亚胺)作为氧化还原介质进行选择性氧化,产生了α-酮酸,这些α-酮酸可以直接泵入阴极室(见图11B)。135 随后在钛阴极上的还原胺化反应能够生成氨基酸作为最终产物。这种级联策略利用水作为氢源,并产生多种氨基酸(例如甘氨酸、丙氨酸和亮氨酸),具有不错的产率和法拉第效率。
除了在阳极室内进行的有价值氧化反应之外,这不可避免地需要将产物从电解液中分离出来,现场卤化可以与现场氢化结合,使这两个期望的反应能够在电化学电池之外的独立反应器中进行。如图12所示,通过利用氢气(H2)和卤素气体(即Br2和Cl2)从水性电解液中的相分离,可以在阴极处由水还原产生的氢气被输送到与阴极室物理隔离的反应器中进行所需的氢化反应。同时,在阳极室内进行的卤化物氧化将生成卤素气体,以在另一个独立反应器中进行目标卤化反应。与其利用水氧化作为反反应只产生氧气这种低价值产物,现场卤化不仅将卤原子引入更有价值的氧化产物中,而且所需的电压输入远低于OER。由于氢化和卤化反应都在电化学电池外的独立反应器中进行,这种现场氢化/卤化策略不仅提高了原子经济性和能量效率,还绕过了从电解液中分离能源密集型产品的步骤。
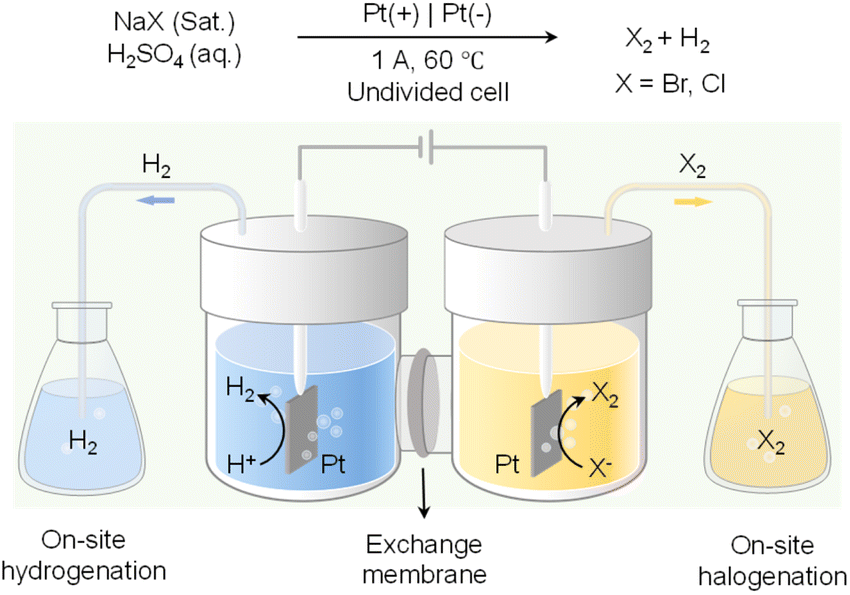
图12 配对电解,包括现场氢化与现场卤化(X:Br或Cl)。
3.4 使用钯膜电极的氢化反应
钯对氢气吸收的现象最早由Graham在1866年观察到,最近的研究表明,当钯被用作阴极时,吸收的H可以推动H/Pd比例超过0.9,其中水是氢源。由于钯面心立方晶格中氢原子的优异渗透性,Iwakura等在1996年报告称,钯箔可以作为阴极,在电化学电池外的腔室中进行苯乙烯的氢化反应生成乙苯(见图13)。137 与传统的电催化氢化不同,Iwakura的设计使用了钯箔作为膜电极(Pdm),起到双重作用:一方面作为阴极通过水的还原产生吸附的H*(Pd + xH+ + xe- → PdHx),另一方面作为物理屏障将氢化室(PdHx + R → PdHx−1 + R–H)与电化学电池分开。继这一开创性工作之后,许多底物已被探索用于使用Pdm作为膜电极和水作为氢源的氢化反应。138 最近,Berlinguette团队进行了几项系统研究,以阐明钯晶体面、共催化剂、载体、溶剂、电池设计和施加电流对各种氢化反应结果的影响,进一步扩大了应用范围,并为使用Pdm的电力驱动氢化提供了深刻的见解。使用Pdm的电力驱动氢化还应用于从烯烃原料中去除丁二烯杂质。
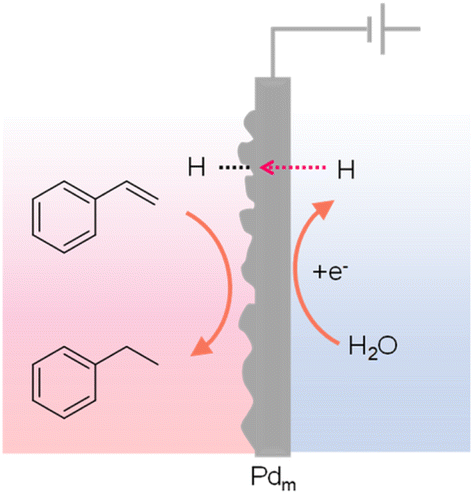
图13 使用钯片电极将苯乙烯氢化为乙苯。137
在大多数使用Pdm的氢化系统中,目标氢化反应通常在阴极Pdm外的某个反应器中进行。受低电位甲醛氧化作为阳极反应的“双重”H2释放启发,Sun等探索了采用Pdm电极作为阳极和阴极,使得在两个Pdm电极外同时进行氢化过程。150 这种“双重”氢化系统使氢化速率加倍,从而提高了整体反应效率。此外,由于甲醛氧化与水氧化相比所需的氧化电位极小,这种双重氢化系统的总电压输入也远小于传统电催化氢化系统。在这种新颖的双重氢化设计中,通过水还原在Pdm阴极产生H*,类似于传统的氢化系统。显著的区别在于阳极侧,Pdm阳极促进甲醛到甲酸盐的低电位氧化,并在Pdm阳极生成H*。这些H*原子不是被氧化成H+,而是可以通过Pdm阳极渗透,在位于阳极室外的独立化学室中催化氢化反应。尽管在Pdm阳极和阴极的电化学过程不同,但两者都有助于H*的产生,有效地将法拉第效率提高了一倍。利用四室装置,当马来酸作为底物时,其氢化产物琥珀酸在Pdm电极相邻的两个外部室中获得。值得注意的是,此过程表现出理论上的最大法拉第效率为200%,即每个经过的电子可以生成两种H*物种,从而将氢化的法拉第效率翻倍。实际上,在施加电流为10 mA时,实现了184%的总法拉第效率。与此同时,与传统需要阳极水氧化的单侧氢化系统相比,电池电压降低了1 V以上。除了烯烃氢化外,炔类底物如4-乙炔基苯胺也可以使用。事实上,当在电解池中使用普通甲醛在D2O中时,在Pdm阴极室外的室中获得了氘化产物,而在Pdm阳极室外的室中产生了普通氢化产物(见图14)。这些结果证实,水是阴极室中的氢源,而甲醛是阳极室中的氢源。简而言之,这项工作证明了同时在不同的化学室中进行氢化和氘化是可行的。
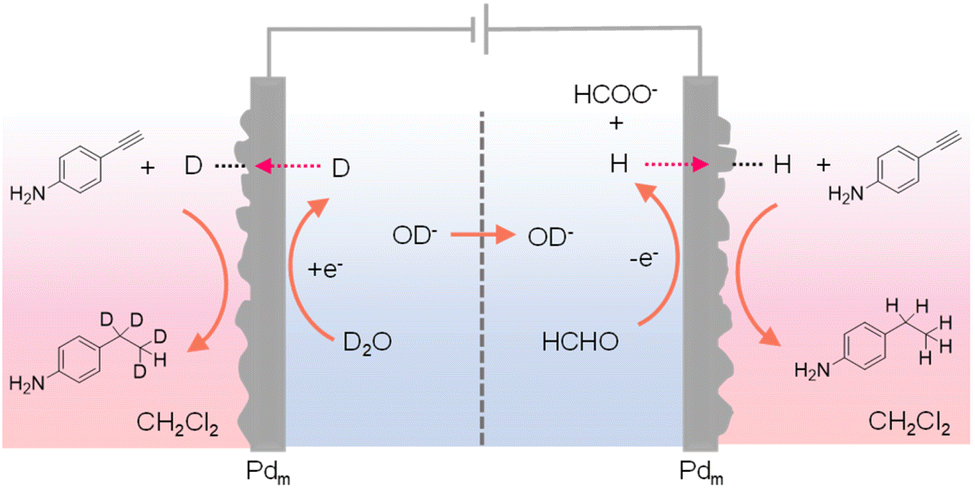
图14 四室装置用于在阳极室外的室中氢化4-乙炔基苯胺,并在靠近阴极室的室中进行氘化。4. 结论与未来展望
在这篇综述文章中,我们深入探讨了水作为氢源在先进有机氢化最新发展中的前景。我们的讨论从比较更传统的氢源与水的优势和局限性开始,随后介绍了几种以水为唯一氢源的代表性电催化氢化反应。尽管这一领域已经取得了巨大进展,但仍存在一些挑战和进一步探索的机会。例如,与热催化氢化相比,目前利用水作为氢源的电催化氢化反应仍然相当有限。预计利用电催化的温和条件,应探索更多多样化和具有挑战性的氢化反应。在此背景下,电力/光驱动的生物催化是一个特别吸引人的方向,尤其是对于不对称氢化反应而言,这还有待更多的关注。
此外,使用重水(D2O)生成氘化化合物和药物分子是一个值得注意的应用。通过将氘引入药物分子,可以通过动力学同位素效应修改它们的代谢轮廓并增强其稳定性。特别是通过D2O电解,应用氢化原理于氘化反应,有望加速氘化分子的开发。事实上,已经报道了一些有前景的电催化系统用于烯烃、醛等的氘化。预计在这一充满希望的领域中将会有更多的研究成果涌现。
总之,这篇综述文章旨在提供利用水作为唯一氢源的电催化氢化最新进展的简要概述。这里强调的进展预示着一个光明的未来,即利用水作为绿色、丰富且廉价的氢源来推动催化反应,朝着可持续和创新的解决方案迈进。
转载本文请联系原作者获取授权,同时请注明本文来自孙学军科学网博客。
链接地址:https://wap.sciencenet.cn/blog-41174-1454469.html?mobile=1
收藏