
引言
随着全球环境问题的日益凸显,人类对清洁能源的需求越来越大,其中氢能作为一种能量密度高、清洁的可再生能源,将成为石油、煤炭等传统化石燃料的理想替代能源。因此,发展氢能经济对于构建低碳能源体系、实现“碳达峰”及“碳中和”目标具有重要意义。目前,氢能主要通过化石能源的分解来制备,但这不仅会消耗大量的不可再生能源,而且还会产生温室气体。相反,电解水制氢技术由于具有操作简单、副反应少、绿色经济等特点成为了未来重要的制氢方式之一。为了降低反应能耗,通常需要开发高活性、高稳定性的阳极析氧(OER)和阴极析氢(HER)催化剂来降低催化反应能垒,降低制氢成本。
对于OER电催化,由于酸性条件下苛刻的反应环境,大多数非贵金属催化剂难以维持正常的运行,因而仍十分依赖Ir基和Ru基等贵金属电催化剂,为了降低成本,亟需维持高活性的同时降低贵金属载量并提高稳定性;而在碱性环境中,Ni基、Fe基等非贵金属电催化剂展现出了更有利的优势,但仍需进一步提高活性和稳定性。对于HER电催化,目前应用最广泛的是昂贵的Pt基材料,为了实现工业化应用,一方面通过调控Pt的电子结构以增强其本征活性,另一方面则需发展高性能非Pt电催化剂。近年来,已经发展了形形色色的具有新结构及高催化性能的电解水催化剂,并已成功应用于质子交换膜电解槽(PEMWE)和阴离子交换膜电解槽(AEMWE)中,展现出了强劲的工业应用前景。本文总结了天津大学胡文彬教授、北京大学马丁教授、中国科学院大连化学物理研究所李杲研究员、德国柏林工业大学P. Strasser教授、阿卜杜拉国王科技大学张华彬教授、深圳大学王振波教授、哈尔滨工业大学于永生教授以及武汉大学罗威教授等课题组在电解水催化剂方面的最新研究成果。
1. Nano Research:高温冲击超声喷雾热解实现高性能电催化剂的制备
因具有高比表面积和独特电子结构等优势,金属纳米颗粒在异相催化领域受到了广泛关注。然而,高的表面能使其在热力学上不稳定,因而在合成和催化过程中,尤其是在高温环境下,很容易发生迁移和团聚。将金属纳米颗粒封装在合适的材料中可防止迁移、聚集等现象,从而提高活性和稳定性。这些封装材料包括无机氧化物、多孔材料和有机大分子等。与这些封装材料相比,碳材料因大的比表面积、高导电性和低成本脱颖而出。碳壳的包覆不仅可增强金属纳米颗粒的催化活性,而且可防止团聚和腐蚀。此外,碳壳还可优化电子结构而不影响气体分子的可及性。尽管已经开发了如化学气相沉积、聚合物涂覆等方法实现了碳壳的封装,但这些方法过程复杂、耗时且可扩展性较差。
鉴于此,天津大学胡文彬教授、陈亚楠教授和中国科学院上海高等研究院曾建荣研究员等人提出了一种高温冲击超声喷雾热解方法实现了金属@碳核@壳结构纳米复合材料的简易和规模化制备,并在OER电催化中展现出优异的活性和稳定性。该方法是目前制备金属@碳核@壳纳米材料最简单有效的方法之一,为高效连续制备高质量纳米复合材料提供了一种有前景的普适性策略。
本文要点:
1)通过葡萄糖和金属氯化物的热解,结合高温冲击和超声喷雾热解,实现了金属@碳核@壳结构纳米复合材料的高效快速制备,该方法可实现对反应体系的精准控温和均匀加热,从而提高了反应效率和产物质量;
2)所制备的Ni@C-40纳米复合材料由片状碳基质包覆的均一Ni纳米颗粒组成,这些纳米颗粒的平均直径约10 nm,碳壳的厚度约2 nm,高导电性的碳壳不仅可防止Ni纳米颗粒的结块或腐蚀,还可作为电催化过程的电子传输通道;
3)在1.0 M KOH电解质中,具有优化碳壳厚度的Ni@C-40在10 mA·cm-2电流密度下具有242 mV的OER过电位,并可在50 mA·cm-2电流密度下稳定地运行超过20小时,其优异的活性和稳定性可归因于Ni纳米颗粒与碳壳之间优化的相互作用;
4)该方法不仅适用于Ni@C纳米复合材料,还可扩展到其他Metal@C材料的制备,包括Ru@C、Pt@C和Co@C等,展示了该方法的良好普适性和可调性。
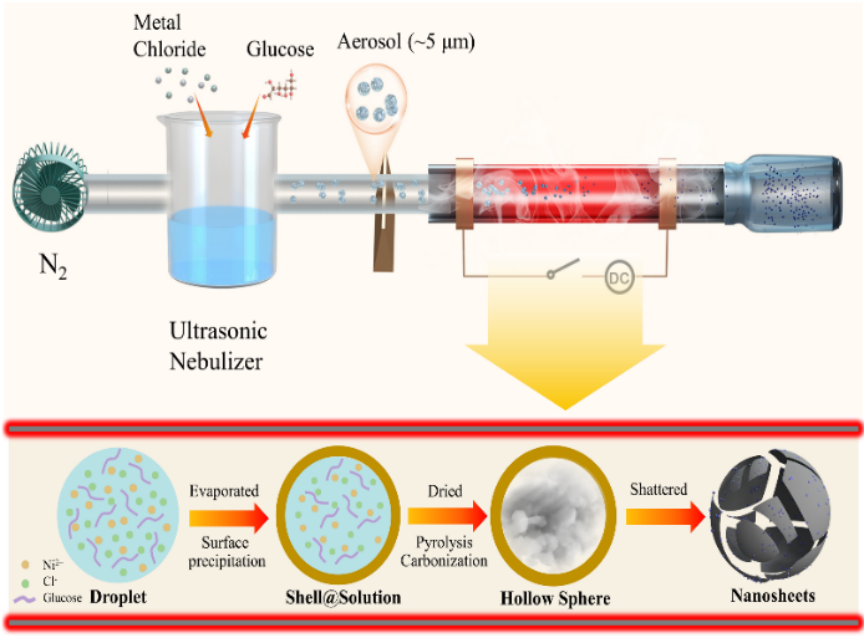
Xiaoyang Wang, et al., Continuous high-temperature rapid nanomanufacturing of electrocatalysts. Nano Res., doi: 10.26599/NR.2025.94907056.

识别二维码访问原文
2. Nano Research:钛滤膜负载二维磷化物异质结构增强安培级海水电解
目前,商业电解水技术十分依赖淡水作为主要来源,但其资源有效。相比之下,海水资源更为丰富,因此以海水为主要来源的海水电解技术将会是未来制备H2的主要发展方向之一。然而,海水中复杂的溶解离子(Na+、K+、Mg2+、Ca2+、Cl-和SO42-等)会限制催化活性和耐久性,特别是竞争性的析氯反应会降低整体海水电解效率。因此,设计和制备高效、高选择性和稳定性的电催化剂对于碱性直接海水电解至关重要。由于优化的电子结构和独特的物理化学性质,过渡金属磷化物(TMPs)表现出了增强的电解活性和稳定性,但仍受到单活性位点的限制。尽管已经通过构筑2D超薄纳米片实现了比表面积和活性位点数量的增加,但差的耐腐蚀性进一步限制了其在碱性海水中的耐久性。
鉴于此,天津科技大学尹振教授、北京大学马丁教授和澳大利亚格里菲斯大学王云副教授等人通过原位生长策略构建了一种新型钛滤膜(Ti-MF)负载的二维磷化物异质结构(NiP2@CoP),由于2D异质结构界面上NiP2和CoP之间丰富的活性位点和强的相互作用,在工业级电流密度下实现了碱性海水中的高性能HER和OER双功能电催化,并可应用于高效工业级海水电解。这项工作创制了极具前景的Ti-MF负载NiP2@CoP催化剂,为工业级直接海水电解技术的商业可行性奠定了基础。
本文要点:
1)钛滤膜具有微尺度多孔结构、高机械强度和强耐腐蚀性等优势,因此被选为电极基底,并通过水热结合磷化处理方法负载了NiP2@CoP异质结构,其具有超薄2D纳米片形貌和丰富的孔结构,丰富的界面位点导致了电荷的再分布,使电子从Co原子转移到Ni原子;
2)在含有0.5 M NaCl的1.0 M KOH电解质中,所制备的NiP2@CoP在500、1000和1500 mA·cm-2电流密度下的过电位分别为425、509和569 mV,优于NiCoP、CoP、NiP2、NiCo2O4和商业IrO2催化剂,在阶梯电流密度下可稳定地运行400小时;
3)NiP2@CoP在碱性海水电解质中还表现出优异的HER性能,在200 mA·cm-2电流密度下的过电位仅150 mV,即使电流密度高达1500 mA·cm-2,其过电位也仅402 mV,同时具有优异的稳定性;
4)将NiP2@CoP分别作为阳极和阴极催化剂组装直接海水电解槽,所组装的NiP2@CoP||NiP2@CoP电解槽仅需要1.90、2.03和2.16 V就可达到200、500和1000 mA·cm-2的电流密度,并可稳定地运行超过300小时。
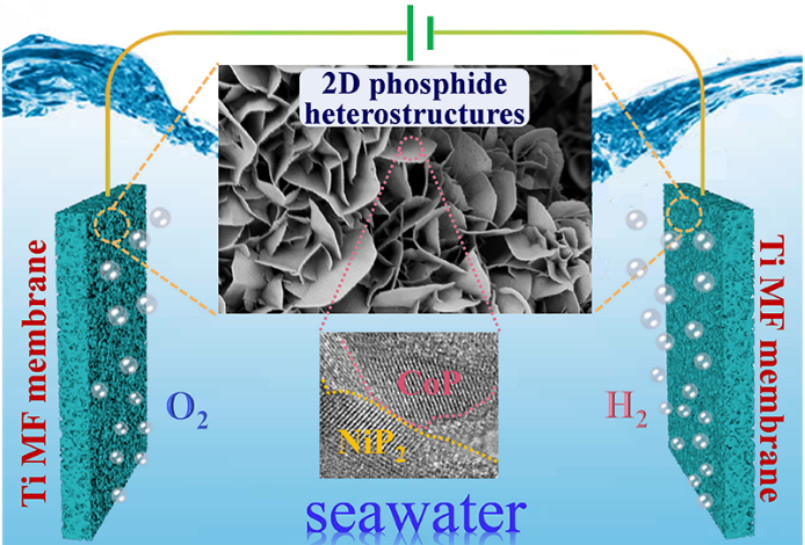
Wenjing Dai, et al., 2D phosphides heterostructures on titanium microfiltration membrane for enhanced ampere-level current density overall seawater splitting.Nano Res., doi: 10.26599/NR.2025.94907061.

识别二维码访问原文
3. Nano Research Energy:SnO2自组装立方纳米笼锚定RuOx团簇实现可持续酸性水氧化
尽管RuO2在高氧化酸性环境中表现出优异的OER活性,但在长时间循环过程中很容易形成可溶性RuO4,降低稳定性。为了提高催化效率并减少Ru的负载,已经提出了包括金属掺杂、缺陷工程、异质结构等策略。其中,在合适的纳米结构载体上生长Ru基团簇是一种有效的策略来增加活性位点的数量。在这些载体中,SnO2因在酸性环境中的优异稳定性和低成本而备受关注。然而,其半导体性质会阻碍Ru的本征活性,影响载体上Ru基材料的稳定性和活性。增加金属氧化物的表面积有利于促进其在特定环境下的催化应用。整合中空结构和壳状纳米结构可有效增加表面积从而防止聚集,同时有助于促进反应中间体的快速传质。因此,调控SnO2的表面结构有望获得优异的载体来实现RuOx团簇在酸性条件下OER性能的提高。
鉴于此,中国科学院大连化学物理研究所李杲研究员、郭嵩博士和沈阳师范大学赵震教授等人在具有分级多孔中空结构SnO2纳米笼上负载了RuOx团簇(RuOxSnO2),克服了SnO2导电性差的局限,实现了酸性电解质中OER电催化性能的显著增强。这项工作为提高酸性OER活性提供了一条新途径,并为Ru基复合催化剂的未来应用提供了有价值的参考。
本文要点:
1)首先构建了SnO2纳米笼的分级多孔中空结构,其尺寸约1.3-1.6 μm,可有效增加比表面积并促进反应中间体的快速传质,从而克服SnO2导电性差的问题,随后通过水热法负载了尺寸约2.5±1 nm的RuOx团簇,该团簇含有丰富的氧空位;
2)在0.5 M H2SO4电解质中,RuOx-SnO2在10 mA·cm-2电流密度下的过电位仅225 mV,在270 mV电位下的质量活性为6873.4 AgRu-1,达到了商业RuO2的170多倍,并可在50 mA·cm-2电流密度稳定地运行100小时;
3)以RuOx-SnO2作为阳极催化剂,商业Pt/C作为阴极催化剂所构建的PEMWE电解槽具有优异的耐久性,在50 mA·cm-2电流密度下可保持至少100小时,即使在100 mA·cm-2电流密度下也可稳定地运行30小时;
4)RuOx-SnO2高的本征OER性能主要归因于具有丰富氧空位的RuOx团簇尺寸的减小,增加了催化活性位点密度,并通过密度泛函理论计算进一步提出了Ru5c-Ov的双活性位点机制,即五配位表面Ru位点(Ru5c)和氧空位(Ov)之间的适度表面迁移加速了*O→*OOH速率决定步骤的反应动力学,从而实现了高的OER性能。
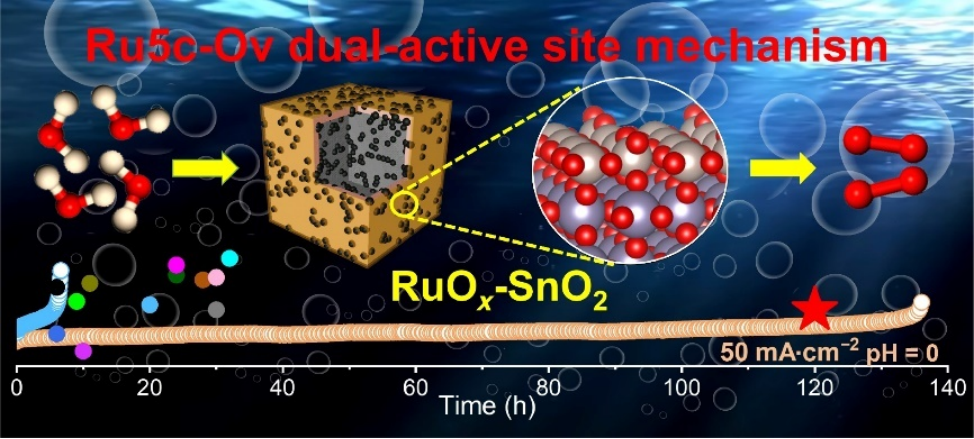
Jingjing Zhang, et al., RuOx clusters anchored on self-assembled SnO2 cubic nanocage for boosting sustainable acidic water oxidation. NanoRes. Energy, doi: 10.26599/NRE.2024.9120140.

识别二维码访问原文
4. Nature Catalysis:具有氧化还原活性Ni-O配体的NiX(X=Fe、Co、Mn)催化层实现高性能阴离子交换膜水电解槽
阴离子交换膜水电解(AEMWE)技术可整合质子交换膜水电解(PEMWE)和液体碱性水电解(AWE)的优势,包括电堆尺寸小、启停速度快以及催化剂成本低等。因此,AEMWE技术成为了大规模可持续产氢的理想选择。近年来,已经实现了各种高导电且稳定的AEM,并在半电池体系下表现出了良好的性能。尽管使用高Ir负载(超过0.5 mgIr·cm-2)的AEMWE阳极可获得安培级电流密度,但对非Ir基AEMWE来说仍是一项巨大挑战。因此,实现高性能、非Ir基且稳定的AEMWE电解槽的设计,并在接近2 V的电池电势下提供>5 A cm-2的电流密度仍是一项巨大的技术挑战。
鉴于此,德国柏林工业大学P. Strasser教授等人报道了一类基于NiX(X=Fe、Co或Mn)层状双氢氧化物(LDH)催化层的非Ir基AEMWE电解槽的设计、组装和膜电极分析,实现了接近酸性PEMWE电解槽的极化特性和产氢速率,在<2.2 V电位下实现了超过5 A·cm-2的电流密度,并结合实验和理论计算阐明了光谱O K边缘特征与Ni基LDH催化剂OER性能之间的相关性。这项工作从分子水平到技术层面上进行了全面考察,通过氧化还原活性Ni-O物种和新型的催化层制备技术实现了与PEMWE技术相媲美的AEMWE电解槽,为更具成本效益和可扩展的产氢技术提供了一条新思路。
本文要点:
1)通过溶剂热法制备了NiFe LDH粉末薄膜作为OER催化剂,发现NiFe LDH具有比NiFe、NiCo和NiMn LDH更高的OER活性,其具有比商业IrOx和NiFeOx更优越的稳定性,这主要归因于NiFe LDH的α和γ相之间的可逆相转变,此外,还发现吸附质电容而非表面积是影响OER反应性的关键因素;
2)通过恒电位原位X射线吸收光谱追踪了Ni和O原子的化学态变化,发现活性的γ-LDH相形成与结构转变密切相关,其涉及层间阴离子喷射和阳离子注入;
3)密度泛函理论计算揭示了NiFe LDH中529eV特征与从非活性α相到催化活性γ相转变的关系,表明该特征与OER活性密切相关;
4)采用湿膜棒涂技术在AEM上直接涂覆催化剂,使用PTFE内箔和粘合剂外箔稳定涂层,制备了具有NiFe LDH阳极的CCM基MEA,在80°C时,在1.8V下可达2A·cm-2电流密度,且在5.5A·cm-2时仍保持动力学和欧姆损耗的可控性;
5)与PEMWE相比,所制备的非Ir基AEMWE电解槽的电位在4A·cm-2时降低了150mV,显示出成本效益,在1A·cm-2下可稳定运行超过110小时,60和80°C下每小时电位增加分别仅600和613µV。
M. Klingenhof, et al., High-performance anion-exchange membrane water electrolysers using NiX (X = Fe,Co,Mn) catalyst-coated membranes with redox-active Ni-O ligands. Nat. Catal., doi: 10.1038/s41929-024-01238-w.

识别二维码访问原文
5. Nature Communications:孤立Ru位点修饰Co3O4的局部压缩应变提高抗腐蚀性以实现高效酸性析氧电催化
在耐酸载体上均匀分散单个贵金属原子是解决酸性OER电催化剂成本高、局限性大等问题的有效策略。在各种耐酸载体中,钴氧化物(如Co3O4)是一种特别有希望的铱氧化物候选替代者,这是因为即使在强酸环境中施加高电势(>2V),Co仍是热力学稳定的。然而,由于钴氧化物在酸性OER条件下的抗腐蚀性能较差,因此在高电流密度或电势下实现长期运行仍具有挑战性。虽然单原子位点修饰的策略可有效解决这个难题,但稳定性增强的本质仍十分模糊。通常,催化剂的局部配位结构对催化性能有重要影响,如晶格畸变、对称性、缺陷等。尽管整体应变效应为建立结构-性能关系提供了有价值的见解,但仍有待探索局部应变的调控方法,这种在局部原子水平上调控的应变效应可能会表现出不同甚至相反的调控机制。因此,需要进一步阐明局部应变调控催化的作用机制。
鉴于此,阿卜杜拉国王科技大学张华彬教授、Luigi Cavallo教授和中国科学院高能物理研究所赵丽娜研究员等人通过在Co3O4基质中引入单原子Ru位点,实现了对局部压缩应变的有效调控,增强了Co3O4在酸性条件下的抗腐蚀性,从而显著提升了酸性OER电催化稳定性,并结合实验表征和理论计算阐明了局部压缩应变对电催化性能的调控机制。这项工作证实了局部应变效应的调控在设计酸稳定OER电催化剂中的巨大潜力。
本文要点:
1)通过阳离子交换策略在中空Co3O4纳米盒表面负载Ru单原子(Ru-Co3O4),Ru的引入缩短了Co-O键键长,实现了局部压缩应变,这一方面源自于原子尺寸较大的Ru原子的引入,另一方面源自于Ru单原子的引入增加了价态,从而改变了相邻Co原子的电子结构;
2)在0.1 M HClO4电解质中,Ru-Co3O4在10 mA·cm-2电流密度下的OER过电位仅252 mV,在30 mA·cm-2的电流密度下运行400小时后,其电位损失可忽略不计,远远优于母体Co3O4催化剂;
3)在酸性OER运行期间,工况条件下的EXAFS分析进一步证实了局部压缩应变的产生,并阐明了OER稳定性的增强主要源自于Co配位环境的稳定,提高了酸性条件下的抗腐蚀性;
4)电解质中的溶解动力学研究表明,Ru单原子的引入显著抑制了Ru-Co3O4催化剂中的金属离子溶解,并通过Ru-O-Co位点协同增强了Ru-Co3O4酸性OER催化剂的抗腐蚀电催化稳定性;
5)从头算分子动力学模拟揭示了Ru-Co3O4中Co元素在酸性条件下增强的抗浸出性能,与Co3O4中Co元素的易溶解特性形成鲜明对比,密度泛函理论计算进一步证实了Co-O结构中的局部压缩应变有助于增强结构和电催化稳定性,并改善相邻Ru位点上的OER动力学,从而实现OER性能的增强。
Shouwei Zuo, et al., Local compressive strain-induced anti-corrosion over isolated Ru-decorated Co3O4 for efficient acidic oxygen evolution. Nat. Commun., doi: 10.1038/s41467-024-53763-8.

识别二维码访问原文
6. Advanced Materials:强金属-载体相互作用和双Cl-排斥层实现超稳定工业级海水电解
为了实现直接海水电解的快速反应动力学,优化活性位点的电子结构并提高电极表面的电子/传质能力至关重要。构建金属-载体相互作用是稳定活性金属、降低贵金属含量和调节催化活性的有效策略。然而,H2/O2气泡的快速形成会阻碍传质,降低催化活性,特别是在工业级电流密度下。为此,构建纳米阵列结构是增强直接海水电解催化剂传质的有效策略。此外,阳极OER还面临着竞争性析氯反应和氯腐蚀等问题。解决这些问题的方法主要有调控局部配位环境、构建保护层及设计富阴离子表面等。在催化剂表面吸附Cl-不仅可排斥游离Cl-以提高OER选择性,而且可调节表面电子结构以提高催化活性,但在工业级电流密度下仍难以完全避免析氯反应。因此,构建双氯排斥层并结合富阴离子表面和氯吸附将是一种有效提高直接海水电解的策略,但尚未被报道。
鉴于此,深圳大学王振波教授和王雷教授等人开发了一种具有强金属-载体相互作用的 Os-Ni4Mo/MoO2微米柱阵列,可用于直接海水电解的双功能电催化剂,加速了催化反应步骤并提高了海水电解效率,其催化稳定性超过美国能源部所设定的技术指标。这项工作为设计高效双功能电催化剂,平衡直接海水电解活性、选择性、稳定性和成本方面开辟了一条新途径。
本文要点:
1)密度泛函理论预测,Os和Ni4Mo/MoO2之间增强的金属-载体相互作用可优化电子结构以增强催化活性;
2)在理论计算的指导下,通过水热结合热处理方法在Ni4Mo/MoO2微米柱上负载了Os纳米簇(Os-Ni4Mo/MoO2),Os簇和Ni4Mo之间形成了金属键,实现了Os和Ni4Mo或MoO2之间强的金属-载体相互作用;
3)在碱性海水电解质中,所制备的Os-Ni4Mo/MoO2在500 mA·cm-2电流密度下的HER和OER过电位分别为113和336 mV,即使在高达1000 mA·cm-2的电流密度下,其HER和OER过电位也仅150和351 mV,并可在500 mA·cm-2电流密度下稳定地运行2500小时,电位衰减率低至0.37µV·h-1;
4)实验和理论分析表明,催化剂的微结构可增强HER和OER电催化传质过程,Os纳米簇不仅通过强金属-载体相互作用优化了电子结构并增强了催化活性,而且实现了优化的*H吸附;
5)对于OER电催化,设计了一个双氯排斥层来抑制析氯反应,这种结构包括催化剂表面原位形成的MoO42-,可通过静电力排斥Cl-离子,此外,强Os-Cl吸附通过静电力形成有效的保护层,进一步降低了电极表面的Cl-浓度;
6)在实际碱性海水电解中,以Os-Ni4Mo/MoO2作为阴极和阳极的电解槽在1.88 V的电位下就可实现500 mA·cm-2的高电流密度,并可稳定地运行超过1000小时,即使膜电极面积达到16 cm2时,电解槽也可在200 mA·cm-2电流密度下稳定运行超过200小时。
Dong Liu, et al., Efficient and ultrastable seawater electrolysis at industrial current Density with strong metal-support interaction and dual Cl--repelling layers.Adv. Mater., doi: 10.1002/adma.202408982.

识别二维码访问原文
7. Angew. Chem.:调控NiFe层状双氢氧化物晶格氧再生增强析氧电催化耐久性
在各种非贵金属基OER催化剂中,NiFe(氧)氢氧化物因其良好的氧吸附/解吸能力而备受关注,然而,Ni和Fe物种在电氧化环境下的溶解极大地限制了长期耐久性的提高。通常,碱性OER途径主要涉及吸附质演化机制(AEM)和晶格氧机制(LOM)。由于热力学限制,AEM的理论过电势限制在0.37 V,而LOM可实现更低的过电势。然而,晶格氧的持续产生往往会导致LOM过程中催化剂的结构坍塌,进一步加速活性物种的溶解。因此,调控晶格氧的再生以弱化活性物种的溶解对于提高Ni基氢氧化物的OER耐久性具有重要意义。此外,加强M-O键是抑制溶解的有效策略,但会阻碍晶格氧的演化。因此,提供额外的氧物种来抵消晶格氧有助于克服晶格氧演化和生成之间的不平衡,但仍存在巨大挑战。
鉴于此,中山大学侯仰龙教授、哈尔滨工业大学于永生教授和杨微微副教授等人提出了一种新型的氧泵策略,以Ni4Mo作为氧泵,实现了NiFe层状双氢氧化物(NiFe-LDH)的晶格氧再生,从而提高了OER催化耐久性。这项工作所提出的氧泵策略为在不影响LOM机制OER催化剂活性的前提下提高耐久性提供了重要指导。
本文要点:
1)通过两步电沉积法在泡沫铜上制备了NiFe-LDH/Ni4Mo,在合成过程中,由于Mo物种在表面的溶解和重构,形成了颗粒-纳米片-颗粒的多级结构,物理表征结果表明,NiFe-LDH纳米片表面形成了丰富的Ni4Mo纳米颗粒,并产生了从Ni4Mo到NiFe-LDH的电子转移;
2)在1.0 M KOH电解质中,所制备的NiFe-LDH/Ni4Mo在10 mA·cm-2电流密度下的OER过电位仅192.5 mV,即使在100 mA·cm-2电流密度下,过电位也仅264 mV,并可稳定地运行60小时,这主要是因为Ni4Mo的引入抑制了Ni和Fe的溶解;
3)原位电化学光谱和理论计算结果表明,由于引入了Ni4Mo,显著增强了对含氧中间体的吸附,加速了晶格氧的再生,从而保证了OER催化活性和耐久性;
4)基于NiFe-LDH/Ni4Mo的AEMWE电解槽在100mA·cm-2电流密度下实现了1.68 V的电解电位,并具有超过150小时的出色耐久性。
Fengyu Wu, et al., Engineering lattice oxygen regeneration of NiFe layered double hydroxide enhances oxygen evolution catalysis durability. Angew. Chem, Int. Ed., doi: 10.1002/anie.202413250.

识别二维码访问原文
8. Angew. Chem.:调控RuO2上的界面水结构和解离实现高效酸性析氧电催化
RuO2是酸性OER的有利替代催化剂,但目前克服其性能限制的调控策略主要集中在优化关键反应中间体(如OH*、O*和OOH*)的结合能上。然而,电荷转移过程和相应的反应动力学还受到双电层结构的影响,但在调控酸性OER动力学方面常常被忽视。理解酸性OER复杂的催化机制及电极-电解质界面环境对于设计具有高活性和耐久性的电催化剂具有重要意义。与碱性条件相比,酸性环境中的水解离需要更高的能量,因此动力学更为缓慢。此外,在酸性介质中,水分子之间相对较强的相互作用还会产生丰富的四配位氢键水(4-HB-H2O)和二配位氢键水(4-HB-H2O),从而形成更强的氢键网络,阻碍了水分子向催化剂表面的迁移,影响解离动力学。因此,阐明催化剂表面的界面水分子在控制表面反应步骤能量和OER动力学的作用是十分有意义的,但仍具有挑战性。
鉴于此,武汉大学罗威教授等人构建了一系列具有可调电子结构和Ru-O共价性的p区金属(Ga、In、Sn)掺杂的RuO2催化剂(Ga-RuO2、In-RuO2、Sn-RuO2),考察了界面水分子结构对酸性OER活性的作用机制。这项工作强调了界面水结构和动态过程在酸性OER中的关键作用,并证明了电化学界面工程在设计高性能电催化剂中的重要作用。
本文要点:
1)通过胶体合成结合热处理法实现了p区金属掺杂RuO2催化剂的合成,纳米颗粒的粒径约4-5 nm,电子从Ru元素转移至p区金属,并引起了Ru的过氧化态,其中Ga原子具有高度的分散性,并以Ga-O键的形式存在于RuO2中;
2)在0.5 M H2SO4电解质中,Ga-RuO2展现出最高的OER活性,在10 mA cm-2电流密度下的过电位仅217.5 mV,低于In-RuO2、Sn-RuO2和RuO2,并且Ga-RuO2可在酸性条件下稳定地运行150小时,而RuO2在很短时间内就会快速失活;
3)密度泛函理论计算结果表明,p区金属掺杂可调控Ru-O键的共价性,并且Ru-O键的共价性与OER催化活性呈现出火山型规律,其中Ga-RuO2表现出优化的Ru-O键共价性,从而使其实现最高的OER活性;
4)工况下的原位红外吸收光谱和理论计算揭示了Ga-RuO2表面游离H2O的局部环境和从4-配位氢键水和2-配位氢键水到游离H2O的动态演变,动态H2O分子的局部环境和氢键网络的调控连接性促进了电子在双电层区的转移,降低了水解离过程的能量势垒,增强了酸性OER动力学。
Liqing Wu, et al., Unveiling the structure and dissociation of interfacial water on RuO2 for efficient acidic oxygen evolution reaction.Angew. Chem. Int. Ed., doi: 10.1002/anie.202413334.

识别二维码访问原文

转载本文请联系原作者获取授权,同时请注明本文来自SciOpen TUP科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3563286-1459692.html?mobile=1
收藏