博文
[转载]Phenomics | 复旦大学公共卫生学院徐望红教授揭示肠道微生物功能群的网络拓扑结构及其在宿主表型关联分析中的优势
||
近日,《表型组学(英文)》(Phenomics)在线发表了复旦大学公共卫生学院徐望红教授题为“Topology of gut Microbiota Network and Guild-Based Analysis in Chinese Adults”的研究论文。
该研究通过构建中国成年人肠道微生物共丰度网络,揭示了菌群的模块化结构特征,并证实了基于“生态功能群”(guild)的分析策略在表型关联研究中的显著优势。
文末点击“阅读全文”可在线阅读文章。

扫描二维码 | 下载PDF原文
论文DOI链接:
https://doi.org/10.1007/s43657-024-00211-8
引用格式:
Fu, J., Yu, D., Zheng, W. et al. Topology of gut Microbiota Network and Guild-Based Analysis in Chinese Adults. Phenomics (2025). https://doi.org/10.1007/s43657-024-00211-8
研究背景
肠道菌群作为人体“第二基因组”,其复杂互作网络与宿主健康和疾病表型关系密切。在微生物组学关联研究中,基于传统分类群(taxon)的分析方法面临显著挑战:同一菌属/种内不同菌株可能呈现截然相反的功能特性,而跨菌属/种的微生物却可能形成紧密协作的功能单元。这种功能异质性在毛螺菌科(Lachnospiraceae)、拟杆菌属(Bacteroides)等核心菌群中普遍存在,导致基于分类学的关联研究常发现不一致的结果。
为克服传统分析方法的不足,学界近年提出"生态功能群"(guild)的概念,即通过共丰度网络聚类,将具有稳定协同关系的菌群作为基本功能单位进行分析。然而,这种分析方法的优势尚未得到充分验证。
研究结果
作者依托上海男性/女性健康队列(SMHS/SWHS),利用2,944名研究对象的肠道微生物16S rRNA V4高变区测序数据,采用SparCC算法,在OTU(operational taxonomic units)水平构建了1,477个OTUs的共丰度网络,发现肠道微生物网络呈现无标度特性,少数核心微生物在网络中占据主导地位,类似于社交网络中的“超级节点”(图1)。
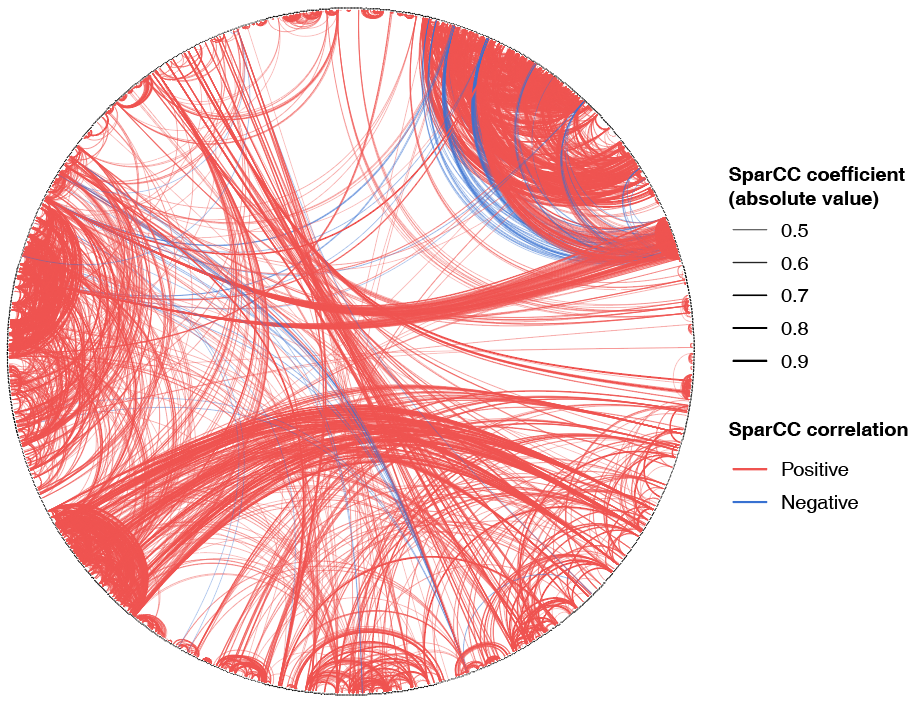
图1 肠道微生物OTU共丰网络
OTU网络节点的接近中心性(closeness centrality)与平均相对丰度(mean relative abundance)、加权节点度(weighted degree)、邻居节点度(neighbor degree)、聚类系数(clustering coefficient)、特征向量中心性(eigenvector centrality)和介数中心性(betweenness centrality)等指标呈显著负相关(图2a);随着加权节点度的增高,细菌OTU越倾向于位于整体群落的边缘(图2b),提示菌群聚集成一个个彼此独立的局部网络,呈现孤立模块化(modularization)特征。
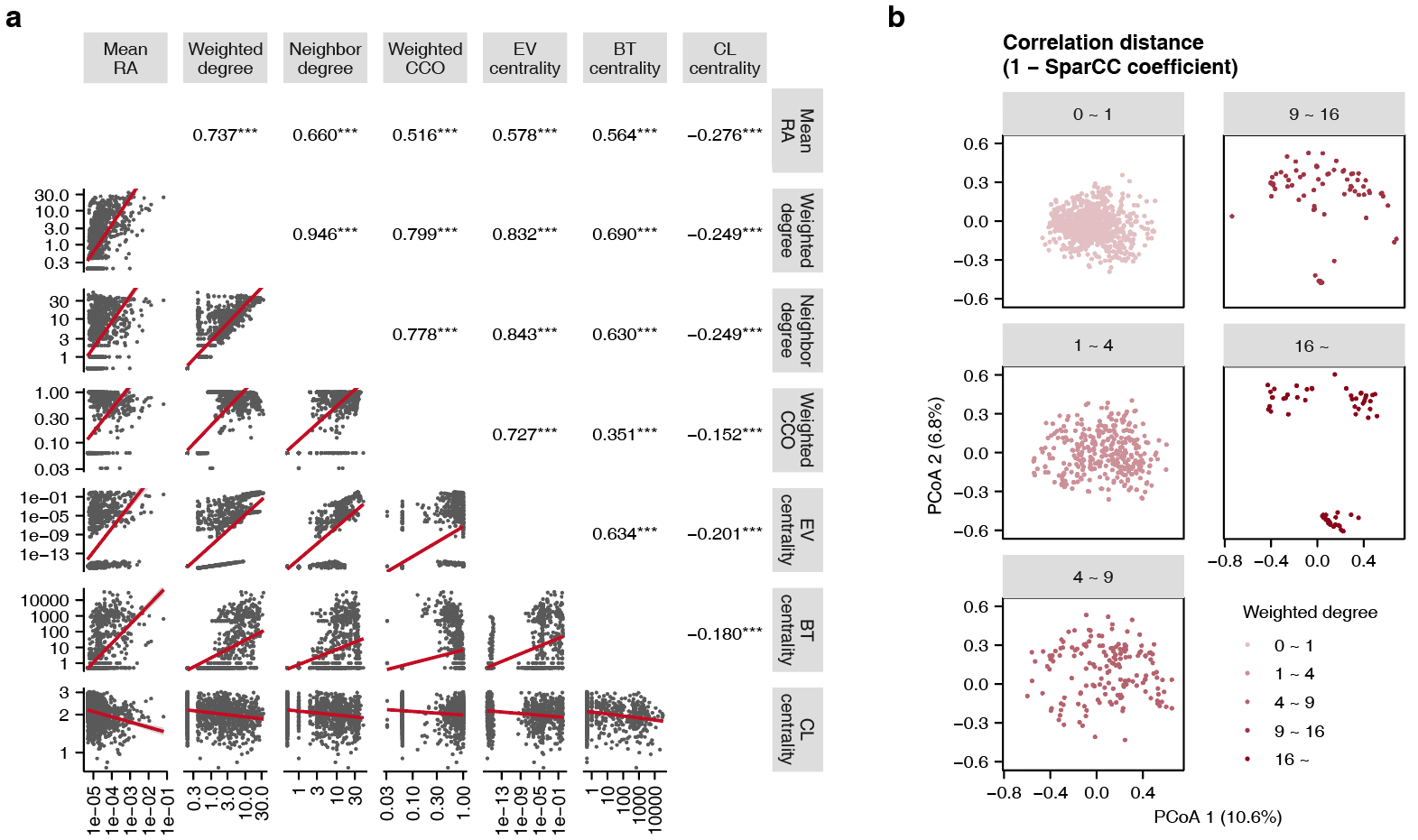
图2 肠道微生物网络的拓扑学特征
这些OTU通过共丰关系共形成130个guild,模块化程度达0.68,高于传统菌属水平的0.60。同一guild内的OTU可来源于不同菌属,而同一菌属的OTU也可分布于不同guild,其中不少OTU未得到准确的分类学注释(图3)。
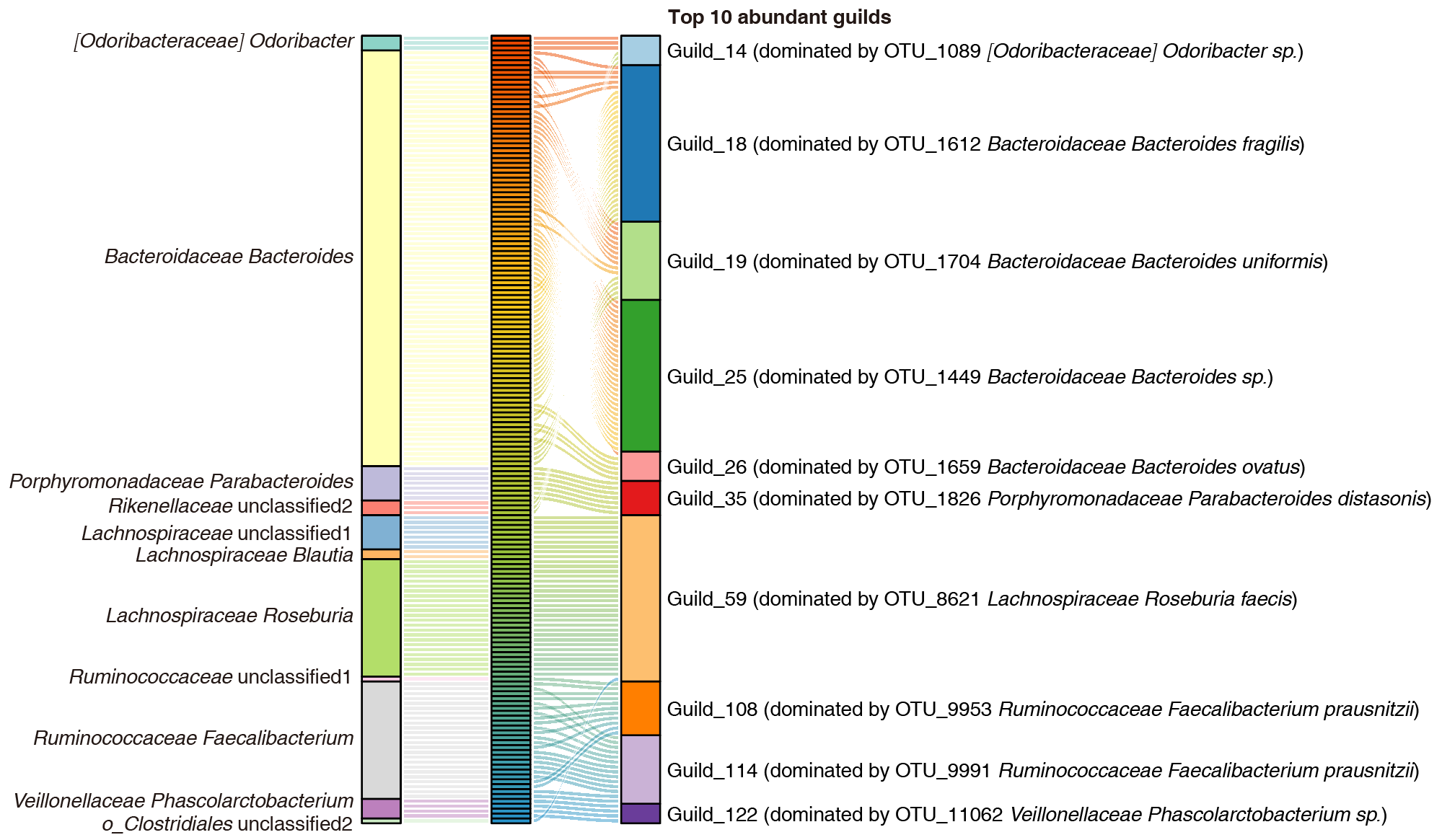
图3 丰度排名前十的功能群的OTU组成
作者在未患和患有常见慢性病的健康和非健康人群中分别构建guild,发现两组人群的guild组成具有高度的相似性(表1),提示在不同健康状况的宿主的肠道环境中,细菌OTU具有类似的共丰行为,guild结构具有稳定性。此外,与菌属水平相比,基于guild的分析能保留更多OTU多样性的原始信息(图4),说明其降维策略的有效性。
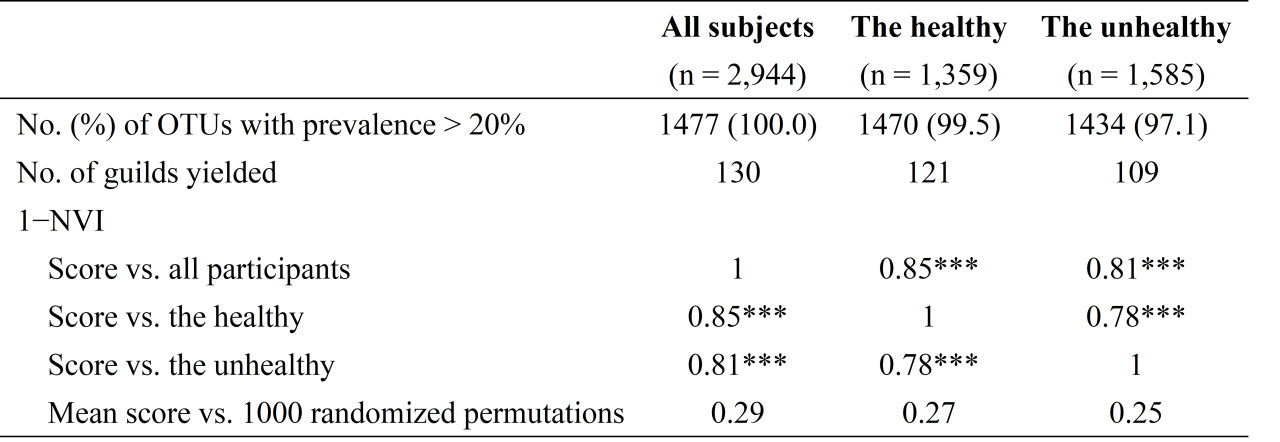
表1 全部、健康和非健康研究对象肠道微生物功能群结构相似性比较
Asterisks represent significance level for one-sample t-test comparing the difference of scores of 1-NVI from real guild structures and random permutations (*: P < 0.05; **: P < 0.01; ***: P < 0.001);
NVI, normalized variation of information.
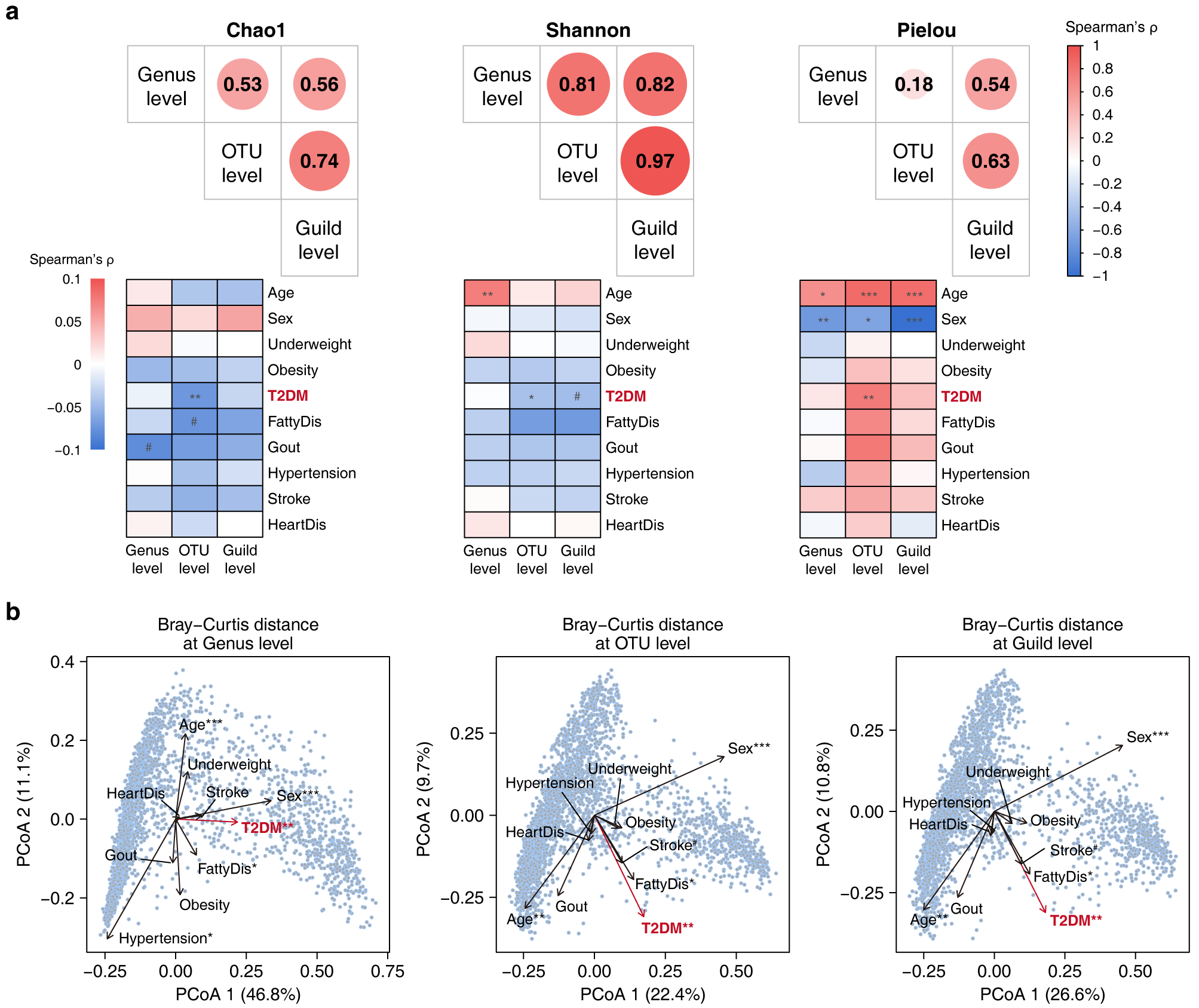
图4 菌属、OTU和功能群水平的微生物多样性特征
作者还比较了基于guild与传统菌属分析方法所得2型糖尿病(T2DM)相关肠道菌群与既往研究结果的一致性。结果显示,本次识别出的13个T2DM相关guild包括了37.0%(17/46)既往已报道的T2DM相关菌属,而传统菌属分析方法仅能识别其中的23.9%(11/46)(图5)。
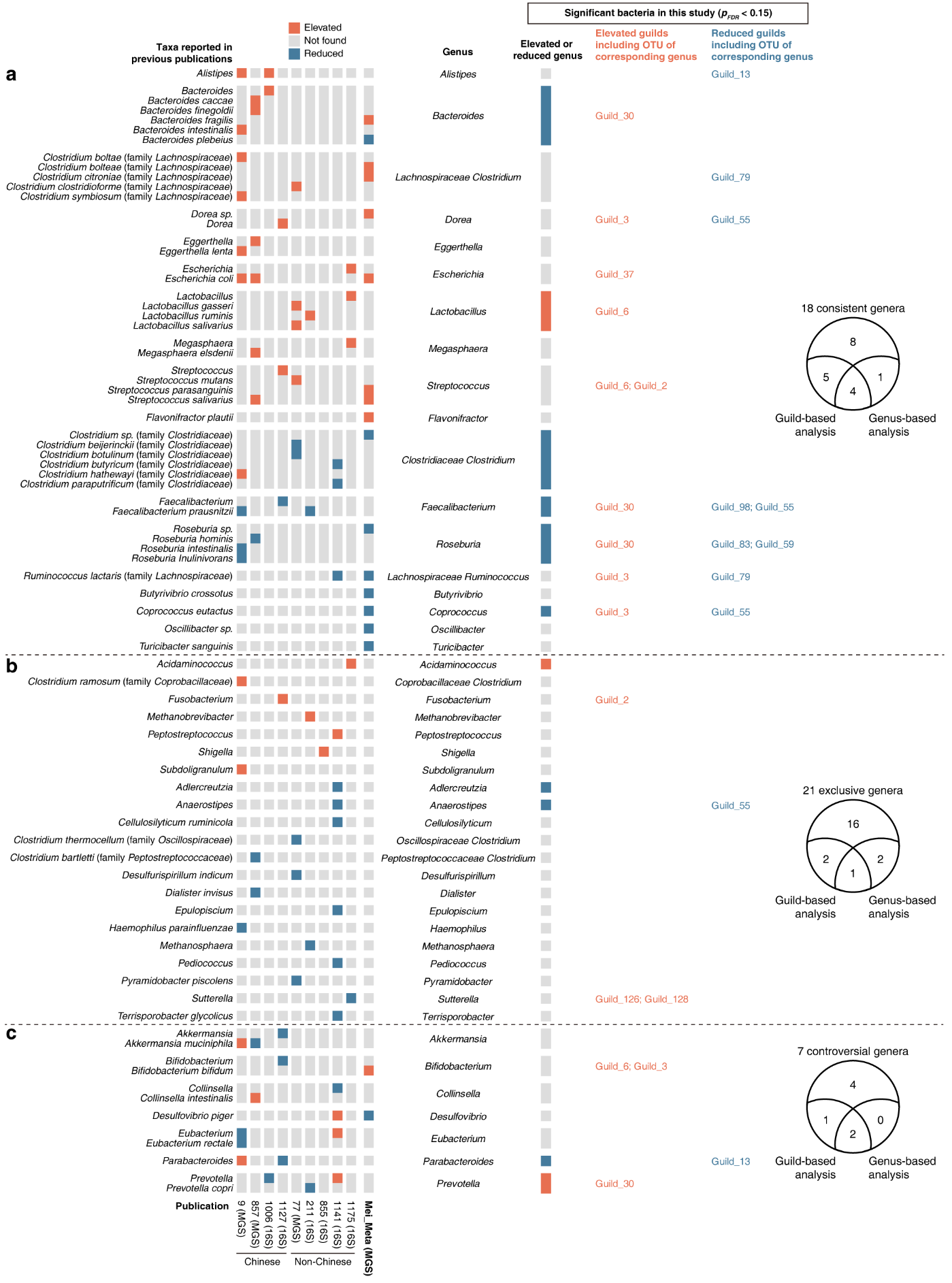
图5 基于功能群和分类群分析发现的T2DM相关肠道微生物的比较
最后,作者总结了基于guild分析的三大优势:①能够区分细菌分类群的功能异质性,可解释部分既往研究的争议性结果;②将未被准确注释的细菌纳入分析,结果更加全面;③考虑了细菌相互作用网络,与宿主表型关联的微生物标志物更具生态学意义。
研究结论
该研究揭示了中国人群肠道微生物guild的模块化拓扑学特征,全面论证了guild作为微生物组研究新范式的生态学合理性及方法学优越性,加深了对肠道菌群与人体健康和疾病表型关联的理解。需要进一步采用多组学研究,深度解析guild的本质和功能特征,推动其临床转化应用及精准干预策略的制定。
Abstract
Gut microbiota with co-abundant behaviors is considered belonging to the same guild in micro-ecosystem. In this study, we established co-abundance networks of operational taxonomic units (OTUs) among 2944 Chinese adults from the Shanghai Men’s and Women’s Health Studies and observed a positive connection-dominated scale-free network using Sparse Correlations for Compositional data (SparCC). The closeness centrality was negatively correlated with other degree-based topological metrics in the network, indicating the isolated modularization of the bacteria. A total of 130 guilds were constructed, with a high modularity of 0.68, and retaining more diversity of OTUs than genus classification. The scores of guild structure similarity for comparisons between all, the healthy and the unhealthy subjects were higher than those derived from randomized permutations, suggesting a robust guild structure. We further used the constructed 130 guilds as the aggregation units to identify gut microbiota that may be associated with type 2 diabetes, and found that the OTUs in 13 significant guilds relevant to diabetes belonged to 17 of 46 (37.0%) previously reported genera (derived from Disbiome database), while only 11 (23.9%) showed different abundances between diabetes patients and healthy subjects in genus-based analysis. Our study reveals modularization of gut microbiota as guilds in Chinese populations, and demonstrates advantages of guild-based analysis in identifying diabetes-related gut bacteria. The analytical method based on microbial networks should be widely used to deepen our understanding of the role of gut microbiota in human health
作者简介
通讯作者

徐望红,复旦大学公共卫生学院教授,博士生导师。长期从事慢性病流行病学研究,曾参与和主持多项国家自然科学基金面上项目和大规模流行病学研究项目,在国内外学术期刊发表学术论文200余篇,其中以第一或通讯作者在BMJ,J Thorac Oncol,Diabetes Care,EClinicalMedicine,Am J Clin Nutr等SCI期刊发表论文60余篇,入选上海市公共卫生优秀学科带头人培养计划,曾获中华预防医学会科学技术奖三等奖、上海市预防医学会科学技术奖三等奖。
第一作者

付炯兴,复旦大学公共卫生学院流行病与卫生统计学博士生,研究方向为营养、衰老、慢性病相关肠道微生物组学研究,以第一作者发表SCI论文3篇。



欢迎投稿
我们诚挚地邀请广大科研人员投稿!
Phenomics官网:
https://www.springer.com/journal/43657
投稿链接:https://www.editorialmanager.com/pnmc/
编辑部邮箱:phenomics@fudan.edu.cn
排版 | 张阿林
撰文 | 付炯兴 徐望红
编辑 | 宋远丽
校审 | 徐望红
https://wap.sciencenet.cn/blog-3558836-1484807.html
上一篇:Phenomics | 复旦大学王瑞团队发布超快速核酸检测技术,可10分钟锁定三种病原体
下一篇:Phenomics | 复旦大学王旭东团队开发新型纳米探针,可揭秘活细胞线粒体有氧呼吸代谢效率
全部作者的其他最新博文
- • Phenomics | 中山大学肿瘤防治中心曾木圣院士团队发表综述:肿瘤诊疗新靶点,整合素α6靶向技术引领精准医疗新时代
- • Phenomics | 上海交通大学医学院张孝勇教授团队开发磁共振成像降噪新方法,有潜力改善脑小血管病的诊断效能
- • Phenomics | 复旦大学倪挺教授团队揭示基因内含子多聚腺苷酸化调控细胞衰老新机制
- • Phenomics | 加拿大阿尔伯塔大学和复旦大学联合开发基于文本、音频和视频的多模态抑郁症检测与评估方法
- • Phenomics | 翟振国/蒋太交/张鹏团队合作开发自适应多基因风险评估模型提升汉族人群静脉血栓栓塞症的风险预测能力
- • 祝贺 | 《表型组学(英文)》成功入选2025年“中国科技核心期刊”