

【文献介绍】
哈尔滨工业大学先进焊接与连接国家重点实验室陈国庆教授团队在《Journal of Materials Science & Technology》刊载的这项研究,通过结合透射电镜(TEM)实验与第一性原理计算,深入揭示了铝合金中第二相界面抑制位错运动的电子机制。该研究利用Materials Studio平台的CASTEP模块,基于密度泛函理论(DFT)模拟了α-Al基体与Al₃Sc、θ′(Al₂Cu)、T₁(Al₂CuLi)三种强化相界面的电子行为,定量分析了价电子密度差异和差分电荷分布。Materials Studio软件的高精度计算能力,使得研究者能够从电子层面直接关联界面键合强度与位错运动阻力,突破了传统显微结构观察的局限性,为理解材料强化机制提供了原子尺度的理论支撑。
本文创新性地从电子输运行为(价电子密度差与差分电荷密度)定量关联界面原子键合强度与位错运动阻力,建立了铝合金第二相强化效应的电子尺度判据。通过Materials Studio软件的CASTEP模块完成以下关键工作:
(1)界面电子结构计算:构建α-Al/Al₃Sc、α-Al/θ′、α-Al/T₁三组界面模型,计算其价电子密度差(Al₃Sc界面高达98%,显著高于θ′的26.8%和T₁的77.2%),证明Al₃Sc界面电子重组能垒最高,导致位错滑移时断键/成键难度最大。
(2)差分电荷密度分析:首次量化界面电子转移强度(单位面积电荷量),发现Al₃Sc界面电荷转移达0.094 e/Ų(θ′仅0.026 e/Ų),剧烈电子交换形成强原子键合,大幅增加位错跨越界面的能量消耗。
CASTEP计算揭示了界面电子传输强度与位错运动阻力的定量关系,证明Al₃Sc因高电子密度差和强电荷转移成为最优强化相。MS软件的高效建模与精确电子分析能力,为从量子尺度设计高性能铝合金提供了不可替代的工具,凸显了计算材料学在揭示微观机制中的核心作用。

【研究背景】
这项研究旨在从电子层面揭示Al基体/第二相界面对位错运动的阻碍机理,众所周知,合金强度的提升核心在于强化相对位错滑移的阻碍,而位错运动(滑移或攀移)的本质是额外半原子面原子与基体原子间化学键的断裂与形成,这一过程直接依赖于电子行为。为此,亟需从电子层面揭示基体/第二相界面对位错运动的阻碍机理为铝合金的强化设计提供更根本的理论基础。
应用模块:CASTEP模块
原文标题:Analysis of interaction between dislocation and interface of aluminum matrix/second phase from electronic behavior
标题中文翻译:从电子行为分析铝基体/第二相界面位错的相互作用
期刊:《Journal of Materials Science & Technology》
DOI:10.1016/j.jmst.2022.07.020
通讯作者单位:哈尔滨工业大学先进焊接与连接国家重点实验室;哈尔滨工业大学 材料科学与工程学院
【理论研究方法】
本研究通过多尺度方法联用揭示了铝合金界面位错阻力的电子机制:基于经验电子理论(EET)的键长差法计算界面价电子密度差(如Al₃Sc/α-Al达98%),结合第一性原理CASTEP模块分析差分电荷密度与原子布居(如Al₃Sc界面电荷转移0.094 e Å⁻²),定量关联原子键合强度;研究创新性地建立电子行为参数(Δρ、单位面积电荷)→位错键重组能垒→宏观强化效应的普适模型,为第二相筛选提供电子尺度判据——例如Δρ>90%的界面(如Al₃Sc)可提升强度>35%。该方法体系可扩展至其他合金系统,推动材料设计从经验试错转向电子计算驱动。
参数设置
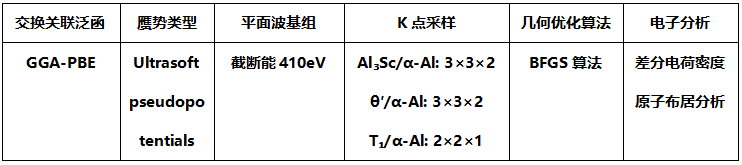
经验电子理论(EET):基于Pauling金属电子理论与能带理论,通过键长差法(BLD)计算共价键长与共享电子对数,量化晶体价电子结构(VES)。
键长计算公式:
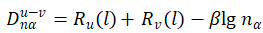
R为单键半径,β为系数。
【研究结果(理论部分)】
1.结构模型的建立
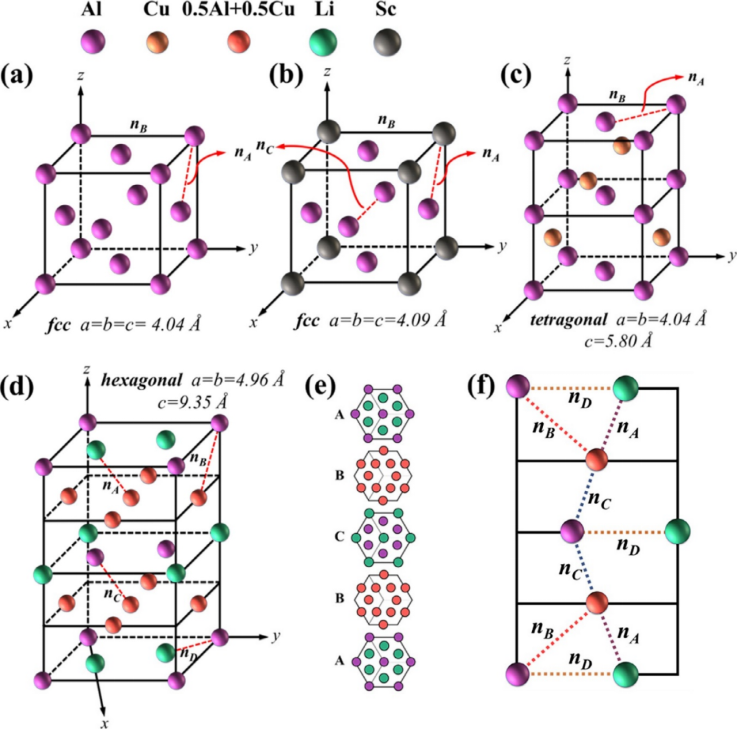
图1. 晶体结构模型
图1为各相的晶格结构:(a)α-Al(面心立方)、(b) Al₃Sc(面心立方)、(c)θ'(Al₂Cu)(四方)、(d) T₁(Al₂CuLi)(六方)的晶格参数及原子排列,(e) 为 T₁相的堆叠顺序,(f) 为 (011) Al₂CuLi 面上的原子排列与共价键。
2.分析不同第二相与α-Al基体的相界面电子密度变化情况
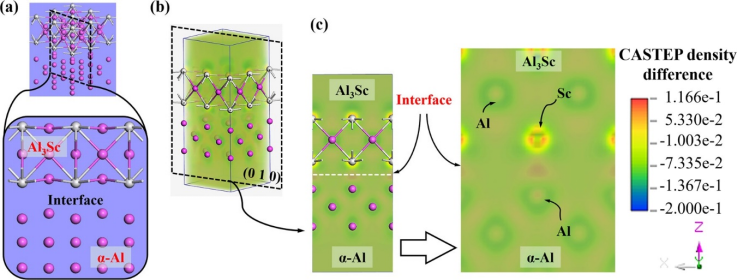
图2. AI3Sc/a-AI界面的差分电荷密度计算结果
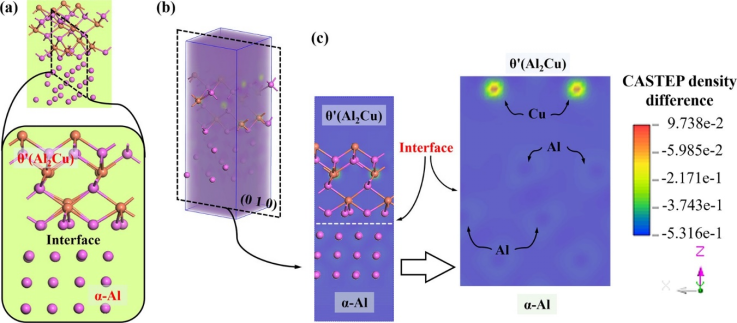
图3. θ′(Al₂Cu)/α-Al界面的差分电荷密度计算结果
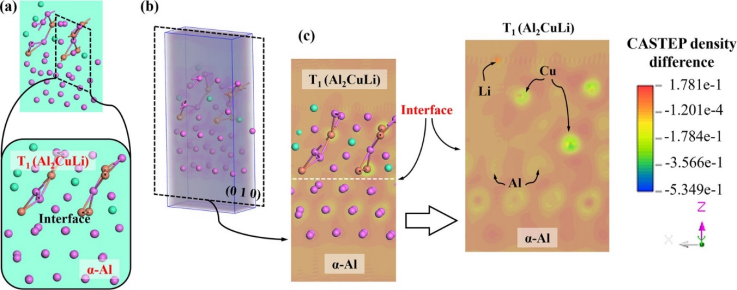
图4. T₁(Al₂CuLi)/α-Al界面的差分电荷密度计算结果
图2~4为Al-Li-Sc合金中的α-Al/Al₃Sc、α-Al/θ′(Al₂Cu)和α-Al/T₁(Al₂CuLi)三种相界的电子输运计算结果(经验电子理论EET和第一性原理CASTEP),将其与透射电镜观测到的结果相关联,说明界面电子转移强度(单位面积电荷量)与对位错运动的阻碍能力直接正相关。对Al-Li-Sc合金中的α-Al/Al₃Sc、α-Al/θ′(Al₂Cu)和α-Al/T₁(Al₂CuLi)三种相界而言,Al₃Sc/α-Al界面因高电子密度差(98%)和强电荷转移(0.094 e Å⁻²)形成强原子键合,大幅增加位错半原子面键断裂/重组难度,导致显著位错塞积和强度提升(457 MPa);而θ′(Al₂Cu)/α-Al界面电子重组容易(电荷密度差26.8%),位错可攀移通过,强化效应较弱;T₁(Al₂CuLi)界面阻碍能力居中。
【研究结论和意义总结与展望】
本研究揭示了铝合金中第二相/基体界面的电子行为对位错阻力的主导机制:界面原子间电子输运剧烈程度(体现为价电子密度差与差分电荷密度)直接决定位错运动的能量壁垒。其中Al₃Sc/α-Al界面因高达98%的电子密度差和0.094 e Å⁻²的电荷转移形成强原子键合,迫使位错断裂/重组键时消耗巨大能量,引发显著位错塞积,使Sc改性合金抗拉强度提升36.8%至457 MPa;而θ′(Al₂Cu)/α-Al界面因电子重组容易(电子密度差仅26.8%)允许位错攀移通过,强化效应较弱。基于此,建议从三方面深化研究:理论层面需建立电子参数-力学性能定量模型,结合原位技术探明变形中界面电子动态演化;设计层面可协同配置高/低电子输运剧烈度的第二相(如Al₃Sc+θ′组合),利用高能界面构筑强度基础,低能界面引导位错有序运动以协同优化强韧性;工艺层面宜通过CASTEP高通量筛选Al₃Zr等新型高电子密度差强化相,并将电子判据拓展至镁/钛合金体系。该"电子行为-界面键合-位错阻力"范式为高强韧合金的电子尺度逆向设计提供了新原理。
End

转载本文请联系原作者获取授权,同时请注明本文来自杨志强科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3536821-1501343.html?mobile=1
收藏