
第一部分:肺炎克雷伯菌的分子分型
1.1 多位点序列分型(MLST)
MLST是一种重要的分子分型技术,常用于研究病原微生物的种群动态、传播规律,监测全球性的高危克隆。截至目前,Pasteur-BigSdb数据库共收录了8081种克雷伯菌的ST分型,揭示了克雷伯菌丰富的遗传多样性。已有的MLST分型研究显示,ST258、ST512和ST11等高流行克隆与多重耐药表型显著相关,主导碳青霉烯类耐药菌的全球传播;而ST23、ST65和ST86等型别则与高毒力表型密切相关,是社区获得性感染,如肝脓肿的主要致病型别。因此,对MLST进行识别与追踪,对于肺炎克雷伯菌的临床感染防控及耐药传播监测具有重要意义。
Institut Pasteur-BigSdb(https://bigsdb.pasteur.fr/)
1.2 血清型(Serotype)
肺炎克雷伯菌的血清型主要包括K抗原分型和O抗原分型。其中,K抗原分型对于肺炎克雷伯菌的致病性至关重要。目前,基于全基因组序列的预测方法已基本取代传统的血清凝集实验,包括以下两种分析策略:
1)基于基因簇结构比对:
通过比对荚膜(cps)或脂多糖(lps)基因簇的整体序列结构进行分型,代表性工具包括 Kleborate、Kaptive 和 Pathogenwatch。其中后两者可以给出基因簇每个基因的匹配相似性,但三种工具均不提供基因在样本基因组中的具体位置信息。因此,在分析cps/lps基因簇结构前,应先基于毒力因子预测或BLAST比对等方法进行定位。
2)基于关键基因的等位基因分型:
通过分析荚膜合成相关基因(如 wzi 或 wzc)的等位基因变异类型来推断血清型,常用平台包括 Pasteur-BigSdb 和 K-PAM。两者均支持基于 wzi 或 wzc 基因序列预测K或O分型。K-PAM 可使用其它更多位点分析,但这两种工具仅能给出血清型别,不提供其它信息。
关键血清型:
研究显示,K1、K2与hvKp密切相关,是引起社区获得性感染的重要毒力血清型。而在ST11-CRKP中,KL47和KL64血清型流行性显著,尤其KL64在全球多个地区呈明显扩张趋势,常与高耐药性和医院内暴发相关。

Kleborate(https://github.com/klebgenomics/Kleborate)
1.3 核心基因组多位点序列分型(cgMLST)
肺炎克雷伯菌的cgMLST分型可以采用以下两种分型方案。
Ridom SeqSphere+ 方案基于2358个核心基因,通常以 ≤15个等位基因差异(Complex Type Distance) 作为阈值来划分克隆复合群(CC),适用于暴发溯源和精细传播链分析。
Institut Pasteur 的方案(scgMLST629_S)则通过多级聚类来界定“亚系(SL)”和“克隆群(CG)”,引入稳定的 cgLIN codes 标识系统,用于与传统MLST分型兼容,支持全球数据整合与标准化命名。

Ridom-SeqSphere+(https://www.cgmlst.org/ncs/schema)
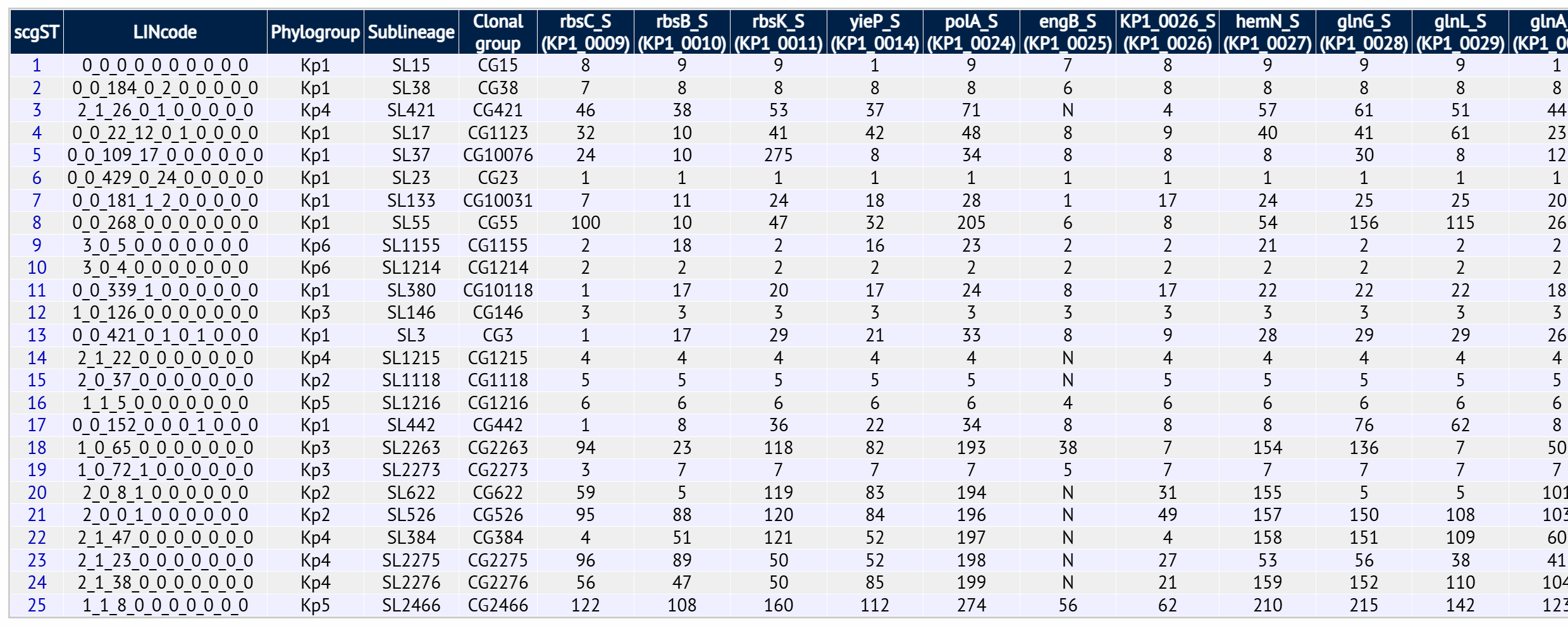
Institut Pasteur-scgMLST629_S(https://bigsdb.pasteur.fr/)
第二部分:肺炎克雷伯菌克隆传播的SNP阈值
以下是与肺炎克雷伯菌克隆传播相关的一些文献,这些已有的SNP阈值可以作为定义肺炎克雷伯菌克隆的参考。
2.1、参考阈值一:≤21 SNPs(欧洲)[1]
2019年欧洲一项发表在Nature microbiology上的多中心研究显示21个SNP是用来确定肺炎克雷伯菌医院clusters(即克隆群)的最佳值,可以最大限度地减少假阳性和假阴性。
作者首先分析了地理环境中成对 SNP 差异的变化。如下图1所示,最小 SNP 差异显示出集中趋势,且从相同医院到相同国家的不同医院,再到不同国家,该模式向右移动,SNP差异总体逐渐增大,即地理和进化距离之间存在着对应关系。
同时,作者采用了类似Aspbury等人的方法,每次从数据集中移除一个菌株作为 “查询样本”(query isolate),其余菌株作为参考数据库,通过比对查询样本与参考数据库菌株的SNP,利用朴素贝叶斯分类计算概率,如果查询样本的最可能来源的医院与其实际采样医院一致,则认为属于院内传播,否则认为是院间传播。
不过需要注意的是,作者选择的样本集中于ST258/512-CRKp,这可能意味着该阈值更适合这一分型的肺炎克雷伯菌研究。
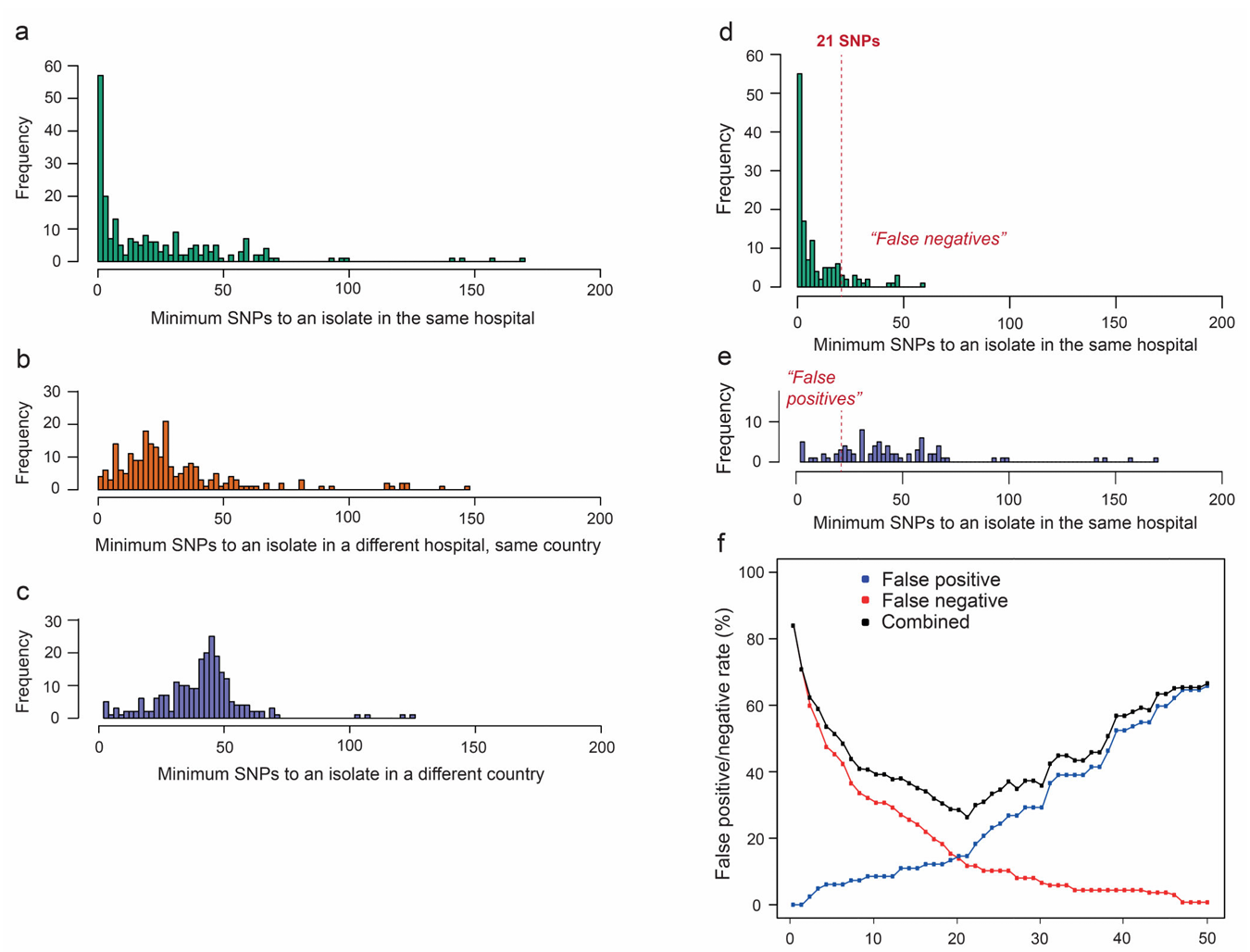
图1 Determination of a SNP cut-off to aid outbreak investigations of ST258/512(https://doi.org/10.1038/s41564-019-0492-8)
2.2、参考阈值二:≤16个SNPs(中国)[2]
本研究通过整合基因组相关性(即核心基因组的成对SNP差异)与患者流行病学关联(即时间与空间的重叠情况),来界定细菌传播簇并调查医院内暴发事件。根据流行病学联系的有无,患者被分为三类:存在流行病学关联(epidemiologic link)、无流行病学关联(non-epidemiologic link),以及附加菌株(additional strain,指同一患者分离出的多个菌株未与其他患者菌株形成聚类)。
系统发育分析显示,研究菌株可划分为三个组别(Group1、Group2 和 Group3)。在优势组 Group1 中,存在流行病学关联的患者菌株与其最近亲缘菌株之间的SNP差异均不超过16。据此,本研究将≤16个SNPs 设定为判定菌株间遗传相关性的最佳阈值。与上文欧洲研究关注于ST258/512克隆不同[1],本研究提出的SNP阈值分析的是国内主流的ST11菌株,该阈值可能更适合ST11菌株的克隆传播分析。
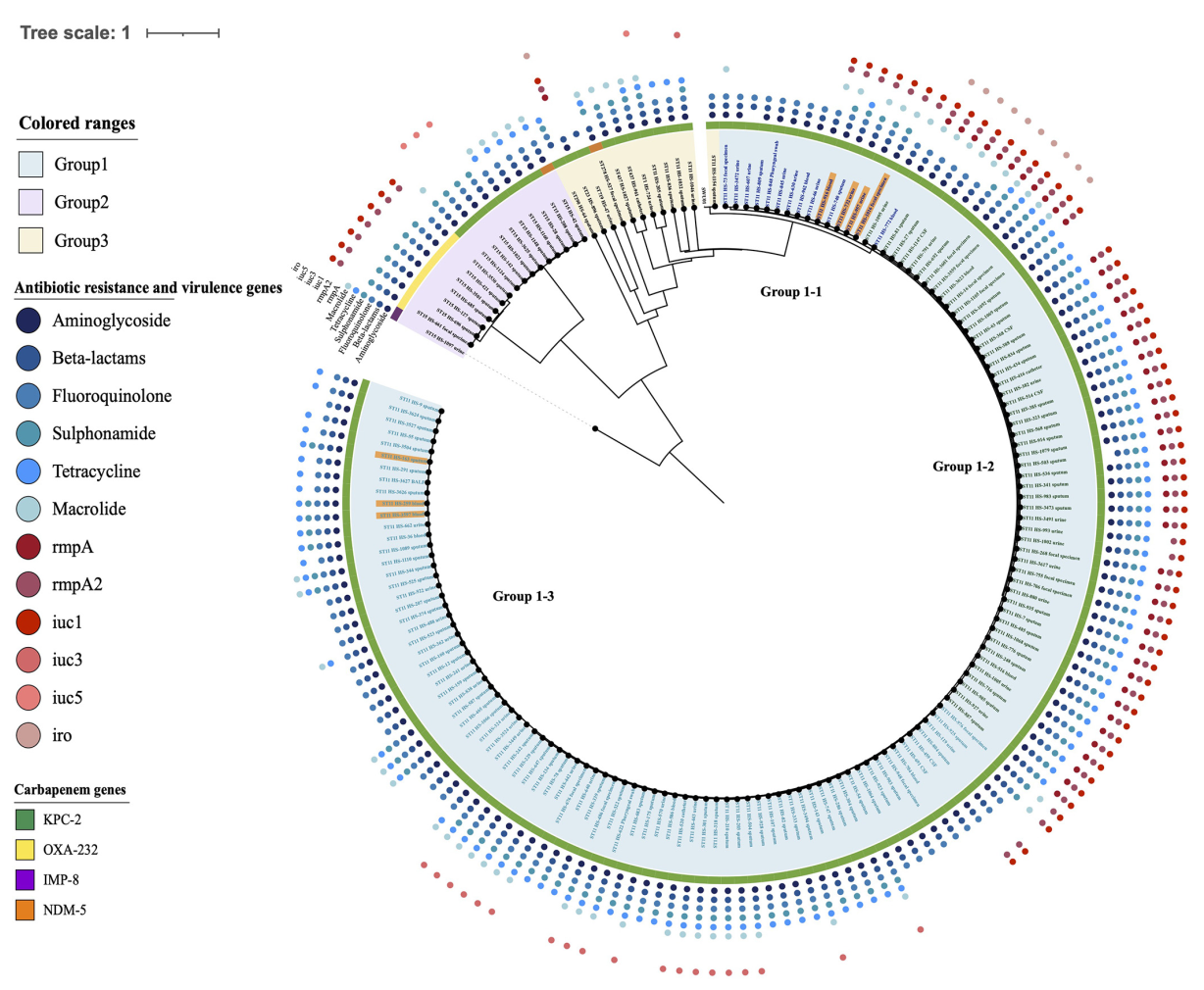
图2 Phylogenetic tree of collected CP-kpn strains(https://doi.org/10.1128/msphere.00143-22)
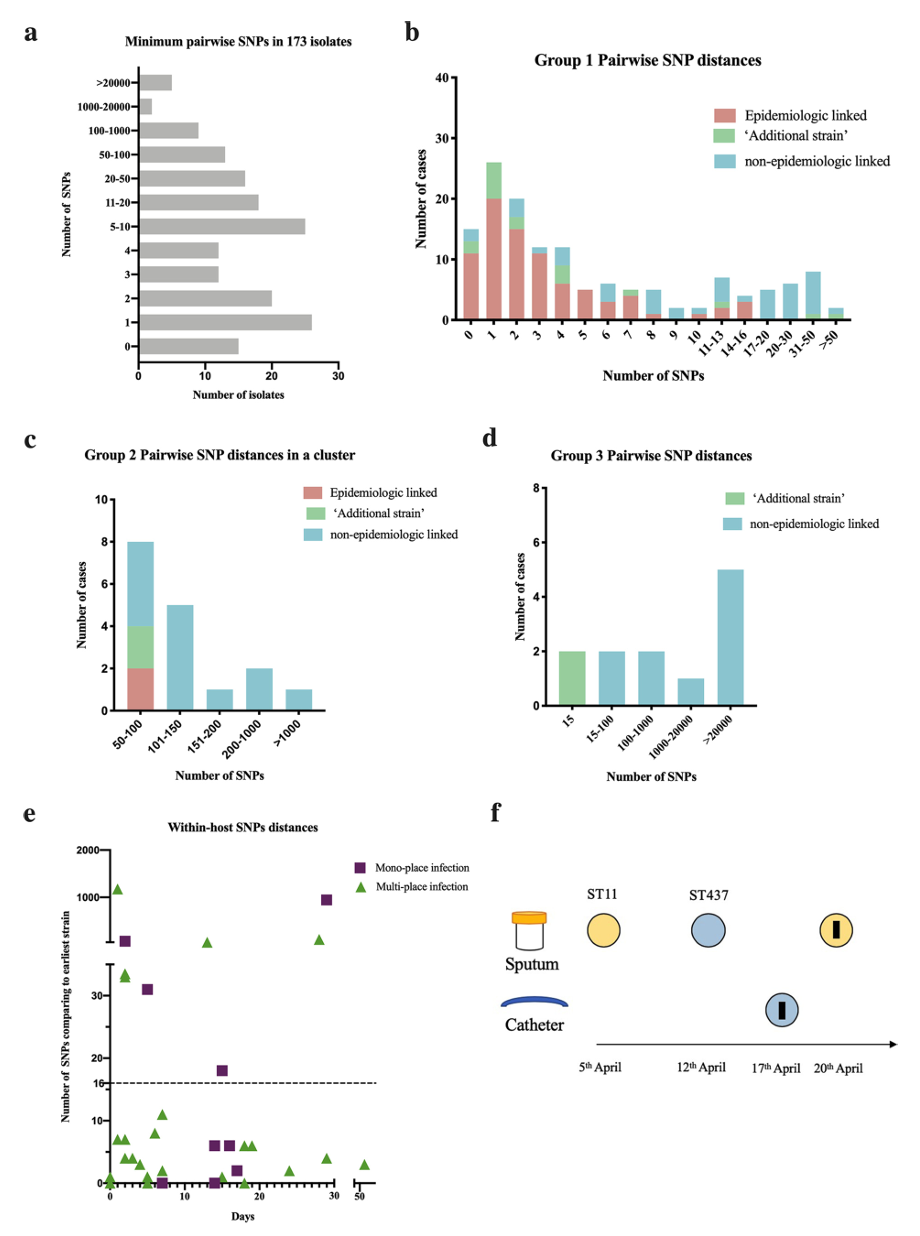
图3 Pairwise SNP distances in 173 CP-kpn strains, clustering distribution, and within-host SNP distances in patients who contributed more CP-kpn strains(https://doi.org/10.1128/msphere.00143-22)
2.3、参考阈值三:≤23个SNPs(澳大利亚)[3]
Sherry, N. L.等人(2019)采用“相同ST型+相同碳青霉烯酶基因+同一物种”的标准划分产碳青霉烯酶肠杆菌(CPE)的亚群(subgroups),进而计算各亚群内的成对SNP距离。分析结果显示,所有在流行病学上被认定为“极有可能”或“可能”的传播对,其成对SNP差异均 ≤23,且95%的传播对SNP≤20。
本文将流行病学联系分为以下4类:
l 极有可能(入院时间和空间上重叠);
l 可能(同时入院但未发现直接联系);
l 不太可能(不同时间入院且未找到直接联系);
l 极不可能(过去4年均有到流行地区的海外旅行史或被不同医院收治)。
方法:首先利用Mashtree比对分离株和RefSeq数据库确定最适参考基因组,随后使用Snippy将测序数据比对至同种同ST的参考基因组以鉴定核心SNPs。基于IQ-TREE(自动模型选择+1000次超快速自举)构建系统发育树,并通过计算亚组内分离株的成对SNP距离分类菌株。针对≥3个分离株的亚组,采用Gubbins(RAxML GTRGAMMA模型迭代10次)屏蔽重组位点,确保进化分析的准确性。
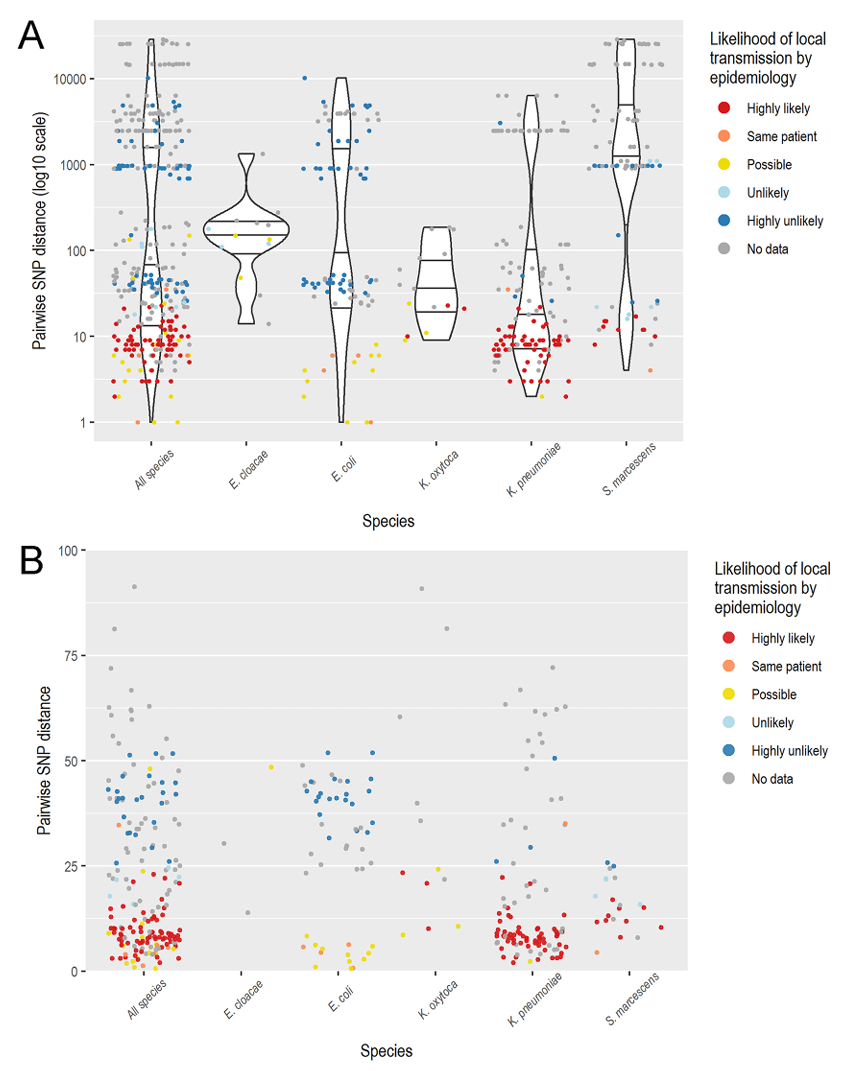
图4 Pairwise SNP distances by species and epidemiologic data(https://doi.org/10.1128/JCM.00573-19)
综合以上三项研究,界定病原菌克隆传播关系的一种策略是:
一、基于分子特征进行初步分组。首先依据MLST、系统发育结构、耐药基因谱等,将菌株划分为不同的群体(如文献二中的Group1或文献三中的subgroups)。利用同一组别内菌株相近的亲缘关系,为后续精细分析奠定基础。
二、结合流行病学联系验证传播事件。在第一步的基础上,进一步整合患者间的流行病学关联信息(如时空重叠情况),通过计算组内菌株之间的核心基因组SNP差异,确定用于界定克隆传播关系的SNP阈值,从而更准确地识别出真正的传播簇。
参考文献
[1] David S, Reuter S, Harris SR, et al. Epidemic of carbapenem-resistant Klebsiella pneumoniae in Europe is driven by nosocomial spread. Nat Microbiol. 2019;4(11):1919-1929. doi:10.1038/s41564-019-0492-8
[2] Zhang Y, Chen C, Wu J, et al. Sequence-Based Genomic Analysis Reveals Transmission of Antibiotic Resistance and Virulence among Carbapenemase-Producing Klebsiella pneumoniae Strains. mSphere. 2022;7(3):e0014322. doi:10.1128/msphere.00143-22
[3] Sherry NL, Lane CR, Kwong JC, et al. Genomics for Molecular Epidemiology and Detecting Transmission of Carbapenemase-Producing Enterobacterales in Victoria, Australia, 2012 to 2016. J Clin Microbiol. 2019;57(9):e00573-19. doi:10.1128/JCM.00573-19
零基础做生信
唯那生物推出的生信云平台服务(https://cloud.mimazi.net)提供了一些与SNP分析、分子分型相关的小工具。用户无需安装软件或配置环境,仅需注册一个账号,找到以下免费小工具,提交对应文件,即可便捷地完成SNP和分型相关的分析,快速获取并下载对应的结果。
1、基于多序列比对结果分析SNP/Indel
2、基于多序列比对文件生成SNP距离矩阵(snp-dists)
3、基于FastA比对文件提取SNP位点(SNP_site)
4、微生物核心基因组比对和 SNP(单核苷酸多态性)检测(Parsnp软件)
5、MLST分析(单个基因组注释)
转载本文请联系原作者获取授权,同时请注明本文来自牛祥娜科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3447233-1503365.html?mobile=1
收藏