
作者:黄必录,胡晓文
摘要:有大量的证据表明,Information Theory of Aging是错误的,并且细胞核和线粒体的DNA损伤也不是导致细胞衰老的原因,我们的研究表明,端粒和rDNA阵列的缩短才是导致细胞衰老的根本原因。
关键词:表观遗传重编程;衰老的信息论;细胞衰老;端粒;45S rDNA
预印本:https://papers.ssrn.com/sol3/papers.cfm?abstract_id=5315671
重要的:
1 衰老的本质是一种遗传程序,而且性成熟后衰老仍然是由程序控制的。
2 要让不变的基因实现程序化表达,要有一种计时物质来驱动。
3 端粒是计时物质的候选者,但有些细胞端粒不会缩短,因此,rDNA阵列候选为第二套计时物质。
1 介绍
以DNA甲基化增减为特征的表观遗传漂移,在小鼠身上比猴子快,而猴子比人快,因此,表观遗传漂移与衰老具有相关性[1]。自从2016年首次在小鼠体内利用山中因子(Oct4, Sox2, Klf4 和C-myc)进行了表观遗传的“部分重编程”的抗衰老研究[2],全球已有大量资本涌入这个方向。例如,投资30亿美元的Altos Labs公司的愿景就是想通过部分重编程来延长人类寿命[3]。因此,澄清表观遗传漂移到底是驱动衰老的原因,还仅仅是由衰老产生的结果,关键到巨额投资的成败。
2023年,David A. Sinclair等人[4]提出了衰老的信息论(Information Theory of Aging,ITOA),所谓的ITOA,就是衰老的表观遗传理论,ITOA描述了衰老过程中表观遗传信息丢失的类型,包括DNA甲基化减少、转录因子失调、非编码RNA、染色质结构改变、组蛋白修饰和丰度等。ITOA理论认为,DNA损伤积累会导致表观遗传信息丢失,而表观遗传信息丢失是驱动衰老的原因。然而,在Cell杂志上已有2篇质疑ITOA的文章[5-6]。
一个正确的理论是不容许存在一个漏洞,并且漏洞越多就越不靠谱。为了推翻ITOA,我们先讨论一个最新的已得到实验验证的衰老理论:Telomere DNA and ribosomal DNA co-regulation model for cell senescence[7],并以此基础指出ITOA的各种漏洞。
2 衰老的本质是一种程序
由于每种生物都有一个相对固定的生长发育、成熟衰老和死亡的时间表,因此,衰老的本质是一种遗传程序。因此,细胞衰老的过程,必然是染色体上的基因群沿着时间轴进行程序化表达的过程,例如,肝细胞在胚胎期主要表达甲胎蛋白,出生后表达白蛋白,到了老年期,白蛋白基因逐渐关闭,转而表达另一些蛋白[8-10]。由于白蛋白是一种不可缺少的蛋白质,因此,测量白蛋白水平可以预测一个人的死亡期[11-12]。
一个生命从受精卵开始,基因表达谱大致可分为早中晚三种模式:早期基因表达谱主要与胚胎发育有关;中期主要与维持健康和生殖有关;晚期主要与破坏正常生理功能有关。因此,衰老和死亡的过程,就是从有益的基因表达谱逐渐转向有害的基因表达谱的过程[13],晚期基因表达谱是导致阿尔兹海默症、动脉粥样硬化和高血压等等的退行性疾病的发病原因。例如,年轻的巨噬细胞主要表达促进新血管形成的,从而增强组织修复的血管内皮生长因子 A 的异构体VEGF-A165A,而衰老的巨噬细胞主要表达抑制血管形成,从而影响组织修复的血管内皮生长因子 A 的异构体VEGF-A165B[14];Clusterin (Clu)在衰老的造血干细胞中表达上调,会促使造血干细胞向髓系分化,导致髓系细胞的产生增加[15]。造血干细胞在衰老过程中,大约有1500个基因表达下调,1500个基因表达上调,主要表现为,对健康有害的应激反应和炎症基因随着年龄增长而上调,而染色质重塑和DNA修复基因随着年龄增长而下调[16]。据此,通常的抗衰老措施就是将某个上调基因进行抑制,某个下调基因进行激活或过表达,但是,这种在代谢层面和信号通路上的干预,只能小幅度延长寿命,而且副作用大,更不可能返老还童。
因此,寻找导致衰老的根本原因不是看什么基因表达上调或下调,而是要找到这些基因为什么会随着年龄的增长上调或下调。
3 性成熟后衰老仍然是由程序控制的
衰老过程是由程序控制的,还是随机损伤积累的结果,这是衰老研究中两派激烈争论的问题。两派的人都同意,生长发育的速度和性成熟的时间是由程序控制的。但是在性成熟后,反对衰老是程序控制的人认为,衰老对个体的作用是负面的,由于自然选择只能对个体起作用,因此,个体不可能发展出并且保持对自己不利的程序,衰老只能是身体受到随机发生的损伤逐渐积累的结果。
但是,生命经过漫长的演化,已经具备了克服各种损伤的防御系统,因此,即使在性成熟后,衰老也不是由随机发生的损伤逐渐积累的结果。以生活在非洲短命的鳉鱼为例,可以说明性成熟后衰老仍然是由程序控制的[17]:鳉鱼产的卵会在干季休眠,并在雨季到来形成水塘后再次孵化。在津巴布韦,那里只有短暂的雨季,雨季过后水塘很快干涸,这里的鳉鱼品系(Nothobranchius furzeri)寿命只有3个月,相当于雨季的长度;莫桑比克的雨季比津巴布韦长4倍,那里的鳉鱼品系(Nothobranchius rachovii)可以活9个月;坦桑尼亚的鳉鱼品系(Nothobranchius guentheri)生活在有两个雨季的地方,寿命可以长达16个月。将这三种鳉鱼放在同一条件下人工饲养,它们寿命的差别仍然存在[18],这说明衰老是由程序控制的,因为随机损伤积累无法解释这三种同一属的身体结构极为相似的鳉鱼寿命为什么差别如此之大,而且正好与雨季的长度相吻合[19]。
4 计时物质驱动遗传程序运行
在一个生命周期里,染色体上大部分基因的序列和拷贝数是固定不变的,因此,要让不变的基因实现程序化表达,就需要一个计时的装置来驱动。由于个体的寿命可以达到百年以上,例如,格陵兰鲨鱼寿命可达400年,因此,候选为驱动遗传程序运行的“计时物质”(相当于沙漏计时器中的沙子)必须非常稳定,没有半衰期。而蛋白质、RNA、线粒体DNA(mtDNA)、以及细胞核DNA和组蛋白的化学修饰,这些都是很不稳定,有半衰期,处于不断降解和补充的动态平衡中,例如,DNA甲基化与去甲基化,组蛋白的乙酰化与去乙酰化都是同时进行的,因此无法形成时间量度,不具备计时物质的属性。也就是说,衰老根本原因不在于RNA、蛋白质、mtDNA和表观遗传修饰上。
“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”认为[7](图1),多拷贝的串联重复序列的端粒DNA阵列和rDNA阵列,完全能满足作为驱动遗传程序运行的“计时物质”的各种要求。端粒DNA阵列和/或rDNA阵列的缩短,可通过P53介导染色体上的基因群沿着时间轴进行程序化表达[20]。从第一性原理看,物种寿命是由端粒DNA阵列和/或rDNA阵列的缩短速率决定的[19]。
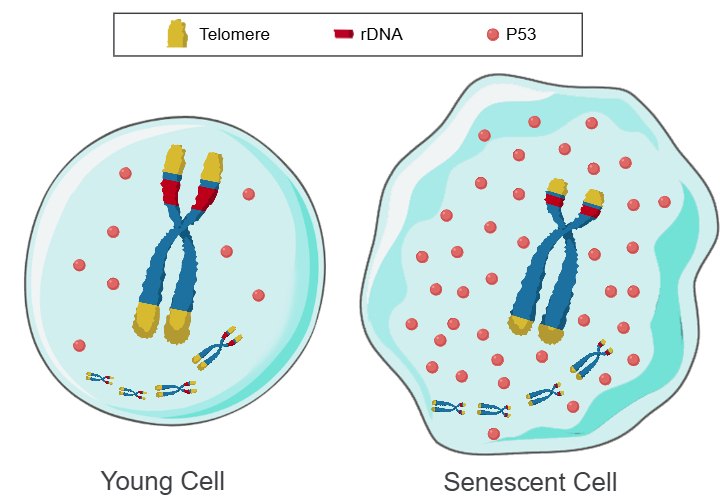
图1:Telomere DNA and ribosomal DNA co-regulation model for cell senescence
a 长阵列的端粒和rDNA,P53迅速降解,细胞年轻。b 短阵列的端粒和rDNA,P53缓慢降解,细胞衰老。
一个理论是否正确,要看这个理论是否具备自洽性,既然端粒DNA阵列和rDNA阵列的缩短是导致细胞衰老的根本原因,是驱动基因群程序化表达的计时物质,那么,在体细胞中损耗掉的端粒DNA和rDNA,在生殖细胞或胚胎的早期细胞中必须要能够补充,否则,生命无法进行世代轮回。幸运的是,已有证据表明,在体细胞中损耗掉的端粒DNA和rDNA,可以在胚胎早期细胞或生殖细胞中得到补充[21-23]。
综上所述,细胞的复制性衰老是由端粒和rDNA阵列缩短导致的。小鼠造血干细胞会因为mTOR1激活导致rDNA阵列缩短[24],而抗衰老药雷帕霉素能通过抑制mTOR1来抑制细胞复制,减缓细胞复制性衰老,延长小鼠寿命。
5 ITOA的漏洞
一个正确的理论是不容许存在一个漏洞,并且漏洞越多就越不靠谱,由于ITOA的漏洞太多,可以确定ITOA是一个错误的衰老理论。
5.1 Telomere DNA and ribosomal DNA co-regulation model for cell senescence证明返老还童机制不是因为表观遗传重编程
我在“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”曾经推测,端粒和rDNA阵列缩短会导致P53水平升高,并且多能重编程返老还童机制是因为端粒和rDNA阵列的大幅度延长[7],为了验证“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”是否是正确的,我们通过敲低小鼠和人类原代细胞中的 45S rDNA拷贝数,结果:衰老标志物P53、P21、P16和SA-β-GAL都按照预期显著上调,端粒长度、细胞活力和细胞传代次数都显著减少。此外还检测了小鼠的衰老细胞和hESC与hiPSC,发现衰老细胞的端粒长度和45S rDNA拷贝数都显著减少了,hESC与hiPSC的端粒长度和45S rDNA拷贝数显著增加了,这些数据有力的证明了hESC和hiPSC的返老还童机制不是因为表观遗传重编程,而是因为端粒DNA阵列和45S rDNA阵列的长度都显著增加了,细胞衰老和Hayflick极限的根本原因是由端粒和 45S rDNA 共同调控的,而且rDNA对衰老的权重大于端粒(未发表的观察)[19]。据此,要想逆转衰老和大幅度延长寿命,替代器官移植和治愈退行性疾病,唯一的方案就是通过增加成体干细胞中的端粒DNA阵列和rDNA阵列的长度,以此使组织和器官返老还童,除此之外,其它的抗衰老措施顶多只能小幅度延长寿命,更不可能返老还童。
5.2 表观遗传的增龄性变化是由端粒和rDNA阵列缩短的结果
由于表观遗传修饰是不稳定的,因此无法成为稳定的“基准时钟(reference clock)”物质,不具备计时物质的属性。例如,通过实验手段逆转与年龄相关的 DNA 甲基化变化,这一效应只能维持15到100 天[25]。因此,在因果关系中,表观遗传的增龄性变化不可能是驱动衰老的原因,就象脸上的皱纹不是皮肤衰老的原因,而是皮肤细胞衰老的结果。因此,表观遗传时钟只能是受端粒和rDNA这种更稳定的“基准时钟”通过P53等介导调校的,例如,端粒能通过改变表观遗传影响基因表达[26];不同长度的rDNA阵列会有不同分布的异染色质/常染色质,从而改变了基因表达谱[27-28];相比衰老细胞,年轻细胞的端粒更长,端粒酶活性更高,TERT被证明与染色质重塑因子相互作用,并调节DNA甲基化[29];DNMT3A水平会随增龄而下调,而该基因缺失的造血干细胞会影响染色体不同区域的甲基化水平,从而改变了基因表达谱[30]。
P53会结合到DNMT1基因的启动子上[31],已知端粒阵列缩短会使P53水平升高[32],我们的实验也证明rDNA阵列缩短会使P53水平显著升高(未发表的观察),因此,随着年龄增长,端粒和/或rDNA阵列逐的渐缩短,P53就会沿着时间轴产生“浓度梯度”,由于P53会与多种基因的启动子和增强子结合,从而使有些基因表达上调,有些基因表达下调,以此驱动染色体上的基因群进行程序化表达。
5.3 直接重编程和部分重编程无法逆转衰老
表观遗传重编程分为多能重编程、直接重编程和部分重编程。在多能重编程中,体细胞失去身份,表观遗传年龄(Epigenetic Age EpiAge)逆转到0岁。由于iPSC和ESC的端粒和rDNA阵列不会缩短,因此,这些细胞不会经历表观遗传老化[33]。当iPSC和ESC分化为体细胞后,端粒阵列很快开始缩短[34],由于表观遗传时钟是由端粒和rDNA阵列调控的[26-28],因此,在端粒和rDNA阵列缩短之时,也是表观遗传时钟开始嘀嗒作响之时[35]。
直接重编程又称“转分化”,将老年成纤维细胞直接重编程为神经干细胞,则保留了衰老的表型,[36],因此,直接重编程不会逆转衰老。
在部分重编程中,这些细胞身份不变,但端粒也没延长或略有缩短,一旦停止表达山中因子,衰老症状又会很快积累,包括EpiAge恢复到重编程之前的状态[37],部分重编程的细胞,生理状态的年轻化发生在表观遗传特征的年轻化之前[38],说明导致衰老细胞暂时性的逆转与表观遗传改变无关,部分重编程是无法长久逆转衰老的。
部分重编程也没有延长野生型小鼠寿命[2],或仅增加野生型小鼠中位寿命12% [39],还不如小分子抗衰老药。
5.4 细胞衰老与DNA甲基化与否无关
细胞越衰老,DNA甲基化水平越低,然而,原始生殖细胞(Primordial germ cell, PGC)的基因组只有少量甲基化的DNA[40],但是,PGC可能和iPS细胞一样具有永垂不朽的潜力;果蝇的DNA很少发生甲基化,秀丽隐杆线虫没有甲基化的DNA。加州大学洛杉矶分校的人类遗传学和生物统计学教授Steve Horvath说,美国国立卫生研究院一直支持我,但说实话,这还不够,因为一些研究人员认为,由于蠕虫没有表观遗传时钟,它怎么会衰老?
5.5 逆转EpiAge对寿命影响不大甚至还会加速衰老
虽然连续补充α-酮戊二酸(AKG)7个月能使EpiAge拨回8年[41],但是,长期摄入AKG的雌性中年小鼠,中位数寿命虽然延长了近16%,但对雄性小鼠的寿命影响不显著[42];生长激素能逆转EpiAge[43],对老年个体生长激素补充实验发现,生长激素可使61-81岁老人肌肉增加皮肤变厚,年轻10-20岁[44],但实际上长期使用生长激素会加速衰老[45-47],一位60岁的老人接受了生长激素释放激素(GHRH)基因治疗后,EpiAge下调了6岁,但端粒年龄却比同龄人老了7个月[48]。提示,一些能逆转EpiAge,增强线粒体功能和促进细胞增殖的抗衰老药,实际上是在透支有限的细胞分裂次数和寿命,EpiAge只能表示新陈代谢率或“代谢年龄”,或者说,EpiAge只能衡量衰老程度,表观遗传漂移并非驱动衰老的因素。
由于端粒DNA和rDNA是很脆弱的串联重复阵列,转录过程很容易使拷贝丢失,而合成蛋白质需要先合成占RNA总量82%的rRNA,因此,生长激素下调EpiAge和缩短寿命的悖论,其实就是促进蛋白质和ATP合成,检测时显示年轻状态,实际上是透支了有限长度的端粒和rDNA的阵列。值得注意的是,很多具有促进蛋白质和ATP合成的所谓的抗衰老药,都有可能透支寿命。例如,增加NAD+的抗衰老药NR能够增加线粒体功能,经最严格的美国国家老龄研究所测试,反而使小鼠寿命缩短3%[49];秀丽隐杆线虫添加抗氧化剂维生素C,线粒体ATP产量增加了2.5倍以上,在治疗后的第8天,脂褐素(LF)含量增加了约18%,同时寿命缩短。相反,能抑制ATP产生的多西环素反而能使秀丽隐杆线虫寿命延长了72.8%。脂褐素含量下降了约50%[50]。
5.6 DNA突变积累不会导致细胞衰老
ITOA理论认为,DNA损伤会导致表观遗传信息丢失,而表观遗传信息丢失是导致衰老的原因。最近有人认为,体细胞突变才是衰老的真正驱动因素,而表观遗传时钟的变化只是体细胞突变导致的细胞功能异常的“副产品”[51]。其实这两种说法都指向DNA损伤、突变和突变的积累是导致细胞衰老的第一因素。
然而,小鼠自然衰老的心肌细胞的细胞核DNA仅有少量的突变[52];细胞核DNA突变积累并不会加速衰老[53];HeLa细胞的细胞核会迅速积累DNA损伤[54],但不影响HeLa细胞的永生性;很多种癌细胞的基因组很不稳定,但癌细胞EpiAge不会老化[55];电离辐射会增加DNA损伤,却不会影响EpiAge[56]。提示,细胞衰老不是由细胞核DNA突变积累导致的。
1958年,Yoshida发现[57],在8小时内,密伊乐藻(Elodoa den.se)有细胞核的原生质体中的叶绿体,经历了衰老和结构破坏的过程,而无细胞核原生质体的叶绿体仍保持绿色和连续积累淀粉;1965年,Wright和 Hayflick把年轻的细胞核植入去核的衰老的细胞质中,结果细胞恢复了年轻,并按年轻的细胞核剩余的分裂次数继续分裂下去。相反,把衰老的细胞核植入去核的年轻的细胞质中,结果细胞出现衰老表型,细胞分裂次数大为减少。这些研究表明,决定细胞衰老的部位不是线粒体,而是细胞核。也就是说,mtDNA突变积累不可能会导致细胞衰老。
研究也表明,小鼠的mtDNA突变和积累不会加速衰老和缩短寿命[58-59]。线粒体超氧化物歧化酶(SOD2)的杂合突变的小鼠,虽然导致氧化损伤和线粒体DNA突变增加,但并没有缩短寿命[60];电离辐射能极大的增加细胞核DNA和mtDNA突变,然而,致死量以下的电离辐射反而能延长果蝇、家蝇、大鼠和小鼠的寿命[61-64];在日本,原子弹幸存者比平均寿命要长[65];植物可以用变异的枝条来选育新品种,就是靠分生组织中的干细胞的基因突变,但植物干细胞的EpiAge并不会因为基因突变发生衰老[66];iPSC可检测的DNA损伤超过70%[67],但iPSC的EpiAge不会衰老[68]。
由于生命在漫长的演化过程中已具备了克服各种损伤积累的解决方案,只是衰老过程遗传程序会逐渐关闭这些解决方案。例如,细胞核DNA突变到一定阈值的细胞会通过细胞凋亡或免疫系统清除掉,突变的mtDNA会通过线粒体自噬和线粒体胞吐(Mitocytosis)途径清除掉[69-70],因此,衰老的分子损伤积累理论也无法解释从寿命只有10几天的秀丽隐杆线虫,到400~500百年的北极蛤和格陵兰鲨,它们之间的寿命为什么有着如此巨大的差距?以及人体内的寿命长达百年的神经元和心肌细胞,与寿命只有几天的白细胞,它们之间的寿命为何差距如此之大?甚至还无法解释Tappel(1973)将自由基清除剂维生素E添加到饲料喂养成年老鼠1年,发现神经元脂褐素确定少见了,但死亡率未见减少。
综上所述表明,包括DNA在内的各种分子的损伤积累不是导致细胞衰老的原因,ITOA是一种错误的衰老假说。
5.7 表观遗传漂移是一种有计划的程序,而非随机损伤积累的结果
是先有鸡还是先有蛋?是DNA先甲基化再导致基因转录沉默,还是基因先转录沉默再导致DNA甲基化?
反义RNA(antisense lncRNA),通常是由编码蛋白质的基因的反义链转录的,并与该基因的mRNA存在序列重叠,占70%的基因均有反义lncRNA,反义lncRNA的转录往往与其基因的正义链转录存在相关性。反义lncRNA的转录时,会使该位点的DNA被DNA主动去甲基化酶TET3识别,从而清除掉该位点的甲基化修饰[71]。因此,是先有转录因子在促进DNA转录,DNA转录时就会去掉甲基,而在缺乏转录因子时,DNA就会被重新甲基化,DNA甲基化修饰的意义似乎是为了防止DNA在缺乏转录因子的情况不受控制的自发转录。提示,表观遗传漂移不是受随机的损伤所驱动。
随着年龄的增长,全基因组DNA甲基化水平会虽然会逐渐下降,但是,有些基因如IFNγ、F3、CRAT 和 OGG,在衰老过程中更容易被甲基化,GCR、iNOS 和 TLR2则更容易被去甲基化[72],而这些基因都与炎症相关。阿尔兹海默症患者神经元中淀粉样前蛋白基因启动子区的甲基化程度也会随着年龄的增加而下降,导致该基因表达上升,从而致使神经系统功能紊乱[73]。因此,衰老或神经退行性疾病不是一种“随机的损伤积累”或“随机的表观遗传信息丢失”,而是自然演化对遗传程序有意的设计。
5.8 生殖细胞返老还童与表观遗传无关
为了生命的世代轮回,在生殖细胞发育的早期,来自父母的表观遗传信息会被全部擦除,然后重新编程[74],同时把时端粒和rDNA时钟拨回到0岁,以便重新开始计时。
小鼠ESC的端粒长度达到110kb,iPSC的端粒长度也从重编程之前的30kb延长到70kb,经过几次分裂达到110kb。但端粒酶敲除的小鼠细胞诱导生成的iPSC端粒依然是30kb,这些iPSC植入囊胚无法生成嵌合小鼠[75]。自然流产的胚胎也显示端粒长度的不足[76]。
我们的检测发现,和衰老的体细胞相比,hESC 和hiPSC的端粒和45S rDNA阵列的长度显著增加了,但是,hiPSC的端粒和45S rDNA阵列的长度明显比hESC更短(未发表的观察),表明hESC 比hiPSC更年轻。据此可解释为什么用iPSC发育的小鼠寿命短于自然小鼠的寿命[77]。总之,这些证据都提示,多能重编程导致的返老还童与端粒阵列和45S rDNA阵列的长度有关,与表观遗传重编程无关。
结语
综上所述,有大量的证据表明ITOA是错误的,并且细胞核和线粒体的DNA损伤也不是导致细胞衰老的原因,我们的研究表明,端粒和rDNA阵列的缩短才是导致细胞衰老的根本原因。
参考文献
[1]Maegawa S, Lu Y, Tahara T, Lee JT, Madzo J, Liang S, Jelinek J, Colman RJ, Issa JJ. Caloric restriction delays age-related methylation drift. Nat Commun. 2017 Sep 14;8(1):539. doi: 10.1038/s41467-017-00607-3.
[2] Ocampo A, Reddy P, Martinez-Redondo P, et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell. 2016 Dec 15;167(7):1719-1733.e12. doi: 10.1016/j.cell.2016.11.052.
[3] Sahu SK, Reddy P, Lu J, et al. Targeted partial reprogramming of age-associated cell states improves markers of health in mouse models of aging. Sci Transl Med. 2024 Sep 11;16(764):eadg1777. doi: 10.1126/scitranslmed.adg1777.
[4] Lu YR, Tian X, Sinclair DA. The Information Theory of Aging. Nat Aging. 2023 Dec;3(12):1486-1499. doi: 10.1038/s43587-023-00527-6.
[5] Timmons JA, Brenner C. The information theory of aging has not been tested. Cell. 2024 Feb 29;187(5):1101-1102. doi: 10.1016/j.cell.2024.01.013.
[6] Yang JH, Hayano M, Rajman LA, et al. Response to: The information theory of aging has not been tested. Cell. 2024 Feb 29;187(5):1103-1105. doi: 10.1016/j.cell.2024.01.014.
[7] Bilu Huang. Telomere DNA and ribosomal DNA co-regulation model for cell senescence. Negative, 2021. 12(3):9-15. doi:10.13276/j.issn.1674-8913.2021.03.003.
[8] [Sigal SH, Brill S, Fiorino AS, Reid LM. The liver as a stem cell and lineage system. Am J Physiol. 1992 Aug;263(2 Pt 1):G139-48. doi: 10.1152/ajpgi.1992.263.2.G139.
[9] Engelhardt NV, Goussev AI, Shipova LJ, et aI. Immunofluorescent study of alpha-foetoprotein (alpha-fp) in liver and liver liver tumours. I. Technique of alpha-fp localization in tissue sections. Int J Cancer. 1971 Mar 15;7(2):198-206. doi: 10.1002/ijc.2910070203.
[10] Camper SA, Tilghman SM. Postnatal repression of the alpha-fetoprotein gene is enhancer independent. Genes Dev. 1989 Apr;3(4):537-46. doi: 10.1101/gad.3.4.537.
[11] Wu CY, Hu HY, Huang N, et al. Albumin levels and cause-specific mortality in community-dwelling older adults. Prev Med. 2018 Jul;112:145-151. doi: 10.1016/j.ypmed.2018.04.015.
[12] Riviati N, Legiran, Indrajaya T, Saleh I, Ali Z, et al. Serum Albumin as Prognostic Marker for Older Adults in Hospital and Community Settings. Gerontol Geriatr Med. 2024 May 7;10:23337214241249914. doi: 10.1177/23337214241249914.
[13] Frenk S, Houseley J. Gene expression hallmarks of cellular ageing. Biogerontology. 2018 Dec;19(6):547-566. doi: 10.1007/s10522-018-9750-z.
[14] Chen M, Chen J, Liu Y, et al. Senescent Macrophages Promote Age-Related Revascularization Impairment by Increasing Antiangiogenic VEGF-A165B Expression. Aging Cell. 2025 Apr 17:e70059. doi: 10.1111/acel.70059.
[15] Sun N, Lin CH, Li MY, et al. Clusterin drives myeloid bias in aged hematopoietic stem cells by regulating mitochondrial function. Nat Aging. 2025 Jun 30. doi: 10.1038/s43587-025-00908-z.
[16] Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010 Apr;45(4):286-90. doi: 10.1016/j.exger.2009.12.010.
[17] Dance A. Live fast, die young. Nature. 2016 Jul 21;535(7612):453-5. doi: 10.1038/535453a.
[18] Genade T, Benedetti M, Terzibasi E, et al. Annual fishes of the genus Nothobranchius as a model system for aging research. Aging Cell. 2005 Oct;4(5):223-33. doi: 10.1111/j.1474-9726.2005.00165.x.
[19] Bilu Huang , Xiaowen Hu. Causality of Aging Hallmarks. Aging and disease. 2025 https://doi.org/10.14336/AD.2025.0541
[20] Andrysik Z, Galbraith MD, Guarnieri AL, et al. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017 Oct;27(10):1645-1657. doi: 10.1101/gr.220533.117.
[21] Marion RM, Strati K, Li H, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009 Feb 6;4(2):141-54. doi: 10.1016/j.stem.2008.12.010.
[22] Liu L, Bailey SM, Okuka M, et al. Telomere lengthening early in development. Nat Cell Biol. 2007 Dec;9(12):1436-41. doi: 10.1038/ncb1664.
[23] Lu KL, Nelson JO, Watase GJ, et al. Transgenerational dynamics of rDNA copy number in Drosophila male germline stem cells. Elife. 2018 Feb 13;7:e32421. doi: 10.7554/eLife.32421.
[24] Xu B, Li H, Perry JM, et al. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet. 2017 Jun 22;13(6):e1006771. doi: 10.1371/journal.pgen.1006771.
[25] Liesenfelder S, Elsafi Mabrouk MH, Iliescu J, et al. Epigenetic editing at individual age-associated CpGs affects the genome-wide epigenetic aging landscape. Nat Aging. 2025 Mar 24. doi: 10.1038/s43587-025-00841-1.
[26] Carlund O, Norberg A, Osterman P, et al. DNA methylation variations and epigenetic aging in telomere biology disorders. Sci Rep. 2023 May 16;13(1):7955. doi: 10.1038/s41598-023-34922-1.
[27] Larson K, Yan SJ, Tsurumi A, et al. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet. 2012 Jan;8(1):e1002473. doi: 10.1371/journal.pgen.1002473.
[28] Paredes S, Maggert KA. Ribosomal DNA contributes to global chromatin regulation. Proc Natl Acad Sci U S A. 2009 Oct 20;106(42):17829-34. doi: 10.1073/pnas.0906811106.
[29] Yuan X, Xu D. Telomerase Reverse Transcriptase (TERT) in Action: Cross-Talking with Epigenetics. Int J Mol Sci. 2019 Jul 7;20(13):3338. doi: 10.3390/ijms20133338.
[30] Challen GA, Sun D, Jeong M,et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011 Dec 4;44(1):23-31. doi: 10.1038/ng.1009.
[31] Peterson EJ, Bögler O, Taylor SM. p53-mediated repression of DNA methyltransferase 1 expression by specific DNA binding. Cancer Res. 2003 Oct 15;63(20):6579-82.
[32] Leri A, Franco S, Zacheo A, et al. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 2003 Jan 2;22(1):131-9. doi: 10.1093/emboj/cdg013.
[33] Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. Erratum in: Genome Biol. 2015 May 13;16:96. doi: 10.1186/s13059-015-0649-6.
[34] Donate LE, Blasco MA. Telomeres in cancer and ageing. Philos Trans R Soc Lond B Biol Sci. 2011 Jan 12;366(1561):76-84. doi: 10.1098/rstb.2010.0291.
[35] Kabacik S, Lowe D, Fransen L,et al. The relationship between epigenetic age and the hallmarks of aging in human cells. Nat Aging. 2022 Jun;2(6):484-493. doi: 10.1038/s43587-022-00220-0.
[36] Mertens J, Paquola ACM, Ku M, et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell. 2015 Dec 3;17(6):705-718. doi: 10.1016/j.stem.2015.09.001.
[37] Gill D, Parry A, Santos F, et al. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. Elife. 2022 Apr 8;11:e71624. doi: 10.7554/eLife.71624.
[38] Zhang W, Qu J, Liu GH, et al. The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Biol. 2020 Mar;21(3):137-150. doi: 10.1038/s41580-019-0204-5.
[39] Sahu SK, Reddy P, Lu J, et al. Targeted partial reprogramming of age-associated cell states improves markers of health in mouse models of aging. Sci Transl Med. 2024 Sep 11;16(764):eadg1777. doi: 10.1126/scitranslmed.adg1777.
[40] Guo F, Yan L, Guo H, et al. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells. Cell. 2015 Jun 4;161(6):1437-52. doi: 10.1016/j.cell.2015.05.015.
[41] Demidenko O, Barardo D, Budovskii V,et al. Rejuvant®, a potential life-extending compound formulation with alpha-ketoglutarate and vitamins, conferred an average 8 year reduction in biological aging, after an average of 7 months of use, in the TruAge DNA methylation test. Aging (Albany NY). 2021 Nov 30;13(22):24485-24499. doi: 10.18632/aging.203736.
[42] Asadi Shahmirzadi A, Edgar D, Liao CY, et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell Metab. 2020 Sep 1;32(3):447-456.e6. doi: 10.1016/j.cmet.2020.08.004.
[43] Fahy GM, Brooke RT, Watson JP, et al. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell. 2019 Dec;18(6):e13028. doi: 10.1111/acel.13028.
[44] Rudman D, Feller AG, Nagraj HS, et al. Effects of human growth hormone in men over 60 years old. N Engl J Med. 1990 Jul 5;323(1):1-6. doi: 10.1056/NEJM199007053230101.
[45] Coschigano KT, Holland AN, Riders ME, et al. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology. 2003 Sep;144(9):3799-810. doi: 10.1210/en.2003-0374.
[46] List EO, Berryman DE, Slyby J, et al. Disruption of Growth Hormone Receptor in Adipocytes Improves Insulin Sensitivity and Lifespan in Mice. Endocrinology. 2022 Oct 1;163(10):bqac129. doi: 10.1210/endocr/bqac129.
[47] List EO, Sackmann-Sala L, Berryman DE, et al. Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR-/-) mouse. Endocr Rev. 2011 Jun;32(3):356-86. doi: 10.1210/er.2010-0009.
[48] Hanley BP, Brewer K, Church G. Results of a 5-Year N-of-1 Growth Hormone Releasing Hormone Gene Therapy Experiment. Rejuvenation Res. 2021 Dec;24(6):424-433. doi: 10.1089/rej.2021.0036.
[49] Harrison DE, Strong R, Reifsnyder P, et al. 17-a-estradiol late in life extends lifespan in aging UM-HET3 male mice; nicotinamide riboside and three other drugs do not affect lifespan in either sex. Aging Cell. 2021 May;20(5):e13328. doi: 10.1111/acel.13328. Epub 2021 Mar 31. Erratum in: Aging Cell. 2022 Nov;21(11):e13672. doi: 10.1111/acel.13672.
[50] Bonuccelli G, Brooks DR, Shepherd S, et al. Antibiotics that target mitochondria extend lifespan in C. elegans. Aging (Albany NY). 2023 Nov 9;15(21):11764-11781.doi:10.18632/aging.205229.
[51] Koch Z, Li A, Evans DS, et al. Somatic mutation as an explanation for epigenetic aging. Nat Aging. 2025 Apr;5(4):709-719. doi: 10.1038/s43587-024-00794-x.
[52] De Majo F, Martens L, Hegenbarth JC, et al. Genomic instability in the naturally and prematurely aged myocardium. Proc Natl Acad Sci U S A. 2021 Sep 7;118(36):e2022974118. doi: 10.1073/pnas.2022974118.
[53]Robinson PS, Coorens THH, Palles C, et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat Genet. 2021 Oct;53(10):1434-1442. doi: 10.1038/s41588-021-00930-y.
[54] Liu Y, Mi Y, Mueller T, et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat Biotechnol. 2019 Mar;37(3):314-322. doi: 10.1038/s41587-019-0037-y.
[55] Issa JP. Aging and epigenetic drift: a vicious cycle. J Clin Invest. 2014 Jan;124(1):24-9. doi: 10.1172/JCI69735.
[56] Kabacik S, Lowe D, Fransen L, et al. The relationship between epigenetic age and the hallmarks of aging in human cells. Nat Aging. 2022 Jun;2(6):484-493. doi: 10.1038/s43587-022-00220-0.
[57] Yoshida Y. On some characteristics of the idioblast in Elodea leaf. J Fac Sci, Niigata Univ Ser. II 1958; 2: 173–178.
[58] Vermulst M, Bielas JH, Kujoth GC, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007 Apr;39(4):540-3. doi: 10.1038/ng1988.
[59] Tamashiro H, Ishikawa K, Sadotomo K, et al. Mitochondrial Respiratory Dysfunction Is Not Correlated With Mitochondrial Genotype in Premature Aging Mice. Aging Cell. 2025 May 2:e70085. doi: 10.1111/acel.70085.
[60] Van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003 Dec 16;16(1):29-37. doi: 10.1152/physiolgenomics.00122.2003.
[61] Lamb, MJ. The effects of radiation on the longevity of female Drosophila subobscura. Journal of Insect Physiology, 1964. 10(3): p. 487-497. doi.org/10.1016/0022-1910(64)90072-1.
[62] Allen RG, Sohal RS. Life-lengthening effects of gamma-radiation on the adult housefly, Musca domestica. Mech Ageing Dev. 1982 Dec;20(4):369-75. doi: 10.1016/0047-6374(82)90104-x.
[63] Caratero A, Courtade M, Bonnet L, et al. Effect of a continuous gamma irradiation at a very low dose on the life span of mice. Gerontology. 1998;44(5):272-6. doi: 10.1159/000022024.
[64] Calabrese EJ, Baldwin LA. The effects of gamma rays on longevity. Biogerontology. 2000;1(4):309-19. doi: 10.1023/a:1026510001286.
[65] Sutou S. Low-dose radiation from A-bombs elongated lifespan and reduced cancer mortality relative to un-irradiated individuals. Genes Environ. 2018 Dec 19;40:26. doi: 10.1186/s41021-018-0114-3. Erratum in: Genes Environ. 2019 Apr 19;41:12. doi: 10.1186/s41021-019-0127-6.
[66] Dai D, Chen K, Tao J, et al. Aging drives a program of DNA methylation decay in plant organs. bioRxiv [Preprint]. 2024 Nov 5:2024.11.04.621941. doi: 10.1101/2024.11.04.621941.
[67] Rouhani FJ, Zou X, Danecek P,et al. Substantial somatic genomic variation and selection for BCOR mutations in human induced pluripotent stem cells. Nat Genet. 2022 Sep;54(9):1406-1416. doi: 10.1038/s41588-022-01147-3.
[68] Koch CM, Reck K, Shao K, et al. Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res. 2013 Feb;23(2):248-59. doi: 10.1101/gr.141945.112.
[69] Lou G, Palikaras K, Lautrup S, et al. Mitophagy and Neuroprotection. Trends Mol Med. 2020 Jan;26(1):8-20. doi: 10.1016/j.molmed.2019.07.002.
[70] Jiao H, Jiang D, Hu X, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. 2021 May 27;184(11):2896-2910.e13. doi: 10.1016/j.cell.2021.04.027.
[71] Canzio D, Nwakeze CL, Horta A, et al. Antisense lncRNA Transcription Mediates DNA Demethylation to Drive Stochastic Protocadherin α Promoter Choice. Cell. 2019 Apr 18;177(3):639-653.e15. doi: 10.1016/j.cell.2019.03.008.
[72] Madrigano J, Baccarelli A, Mittleman MA, et al. Aging and epigenetics: longitudinal changes in gene-specific DNA methylation. Epigenetics. 2012 Jan 1;7(1):63-70. doi: 10.4161/epi.7.1.18749.
[73] Tohgi H, Utsugisawa K, Nagane Y, et al. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999 Jul 5;70(2):288-92. doi: 10.1016/s0169-328x(99)00163-1.
[74] Lee J, Inoue K, Ono R, et al. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development. 2002 Apr;129(8):1807-17. doi: 10.1242/dev.129.8.1807.
[75] Rosa M. Marion,Maria A. Blasco,Han Li,et al.Telomeres Acquire Embryonic Stem Cell Characteristics in Induced Pluripotent Stem Cells[J].Cell Stem Cell,2009,4:141-154.
[76] Huleyuk N, Tkach I, Zastavna D, et al. Can telomere shortening be the main indicator of non-viable fetus elimination? Mol Cytogenet. 2018 Jan 25;11:11. doi: 10.1186/s13039-018-0361-9.
[77] Chen J, Gao Y, Huang H, et al. The combination of Tet1 with Oct4 generates high-quality mouse-induced pluripotent stem cells. Stem Cells. 2015 Mar;33(3):686-98. doi: 10.1002/stem.1879.
转载本文请联系原作者获取授权,同时请注明本文来自黄必录科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3440171-1494269.html?mobile=1
收藏