
衰老标志的因果关系
作者:黄必录,胡晓文
摘要:本文强调衰老机制的因果关系,认为在衰老的十二大标志中,只有端粒缩短才是衰老的原因。“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”认为,端粒和rDNA阵列缩短会通过P53介导各种衰老标志。因此,逆转衰老和大幅度延长寿命的最好办法,就是增加组织中成体干细胞的端粒和rDNA的阵列长度。
关键词:衰老标志;衰老原因;衰老结果;端粒;rDNA;P53
在线发表在《Aging and Disease》:https://www.aginganddisease.org/EN/10.14336/AD.2025.0541
收录pubmed数据库:https://pubmed.ncbi.nlm.nih.gov/40423632/
本文颠覆了传统衰老认知,其核心突破点包括:
理论创新性:
首次提出“端粒和/或rDNA-P53轴”是驱动衰老的核心机制,阐明端粒与rDNA阵列缩短通过P53介导基因时序表达的分子机制。
突破性解释裸鼹鼠长寿现象(端粒延长实为rDNA缩短的代偿)及非分裂细胞衰老谜题(rDNA拷贝数减少主导)。
技术颠覆性:
指明通过 “重建端粒/rDNA阵列” 实现细胞年轻化的干预路径,为抗衰老药物开发提供全新靶标。
首次提出由端粒和/或rDNA-P53轴驱动各种衰老标志物
合理解释传统干预手段(如抗氧化剂)失效原因——未能触及程序性衰老核心驱动器。
产业前瞻性:
为干细胞疗法(移植高拷贝数rDNA的干细胞)及基因编辑技术(靶向延长rDNA阵列)提供理论基石。
介绍
我们每个人都会慢慢变老,然后死亡,而且很多退行性疾病也是由衰老导致的,因此,与其分别解决不同疾病,不如集中解决一个衰老问题。而要解决衰老问题,就要找到导致衰老的根本原因。然而,许多与衰老相关的变化已被观察到并被吹捧为驱动衰老的原因[1]。2023年,发表在《cell》上题为“Hallmarks of aging: An expanding universe”的综述,列出了衰老的十二大标志[2],但是,作者没有对每个标志进行因果关系的论证。因为对于衰老的干预,针对结果,效果不显著,而只有针对原因,才能大幅度延长寿命。
一,细胞衰老的根本原因
通过对代谢层面和信号通路上对衰老的干预,存在延寿幅度小,副作用大,更不可能逆转衰老。因此,在这里,我们必须先探讨一下导致细胞衰老的根本原因,以便接下来对各种衰老标志进行因果关系的论证。
1 衰老的本质是一种程序
由于每种生物都有一个相对固定的生长发育、成熟衰老和死亡的时间表,因此,衰老的本质是一种遗传程序。而且个体水平的程序是由DNA水平的程序所调控的。例如,肝脏中的肝细胞,在胎儿期表达甲胎蛋白(AFP),成年期表达白蛋白,老年期逐渐表达另一些蛋白[3]。因此,一个生命从受精卵开始,基因表达谱大致可分为早中晚三种模式:早期基因表达谱主要与胚胎发育有关,癌细胞的基因表达模式就定格在这个时期,例如,只在胎肝细胞表达的AFP,成年人肝细胞癌变时又会再次表达;中期基因表达谱主要与维持健康和生殖有关;晚期基因表达谱主要与破坏正常生理功能有关,因此,晚期基因表达模式是导致阿尔兹海默症、动脉粥样硬化和高血压等等的退行性疾病的发病原因。因此,基因表达模式的切换可能是导致个体衰老显著的非线性变化的主要原因[4];谷胱甘肽这种内源性的抗氧化剂在45岁之前几乎保持不变,但45岁后就开始出现显著的非线性下调[5],从而促进衰老和增加了蛋白质的氧化损伤,影响蛋白质的流动性和促进退行性疾病慢的发展[6]。
经过漫长的演化,生命已经具备了克服各种损伤的防御系统,因此,即使在性成熟后,衰老也不是由随机发生的损伤逐渐积累的结果。以生活在非洲短命的鳉鱼为例[7],可以说明性成熟后衰老仍然是由程序控制的:鳉鱼产的卵会在干季休眠,并在雨季到来形成水塘后再次孵化。在津巴布韦,那里只有短暂的雨季,雨季过后水塘很快干涸,这里的鳉鱼品系(Nothobranchius furzeri)寿命只有3个月,相当于雨季的长度;莫桑比克的雨季比津巴布韦长4倍,那里的鳉鱼品系(Nothobranchius rachovii)可以活9个月;坦桑尼亚的鳉鱼品系(Nothobranchius guentheri)生活在有两个雨季的地方,寿命可以长达16个月。将这三种鳉鱼放在同一条件下人工饲养,它们寿命的差别仍然存在[8],这说明衰老是由程序控制的,因为随机损伤积累无法解释这三种同一属的身体结构极为相似的鳉鱼寿命为什么差别如此之大,而且正好与雨季的长度相吻合。
既然衰老是由程序控制的,为什么同一物种或同卵双胞胎的寿命存在较大差异?回答:遗传程序的运行速率会受到各种影响化学反应速率因素的影响。
2 衰老的表现是功能的逐渐下降和改变
随着年龄的增长,细胞总蛋白质和ATP的合成速率会逐渐下调,一部分的蛋白质特异性的上调或下调,称差异表达基因(differentially expressed genes,DEGs),从而导致细胞执行各生理种功能的效率逐渐下降和功能改变[9]。成熟肝细胞下调AFP和上调白蛋白就是属于功能的改变,这就是一种调控着个体的生长发育、成熟衰老和死亡的程序。
导致细胞总蛋白质合成速率下降的原因有二个:(1)首先,总蛋白质合成速率下降是由染色质固缩化造成的,因为染色质固缩化不利于DNA转录。而染色质固缩化与组蛋白低乙酰化有关。衰老小鼠肝脏中的组蛋白乙酰化水平下降,影响了肝细胞再生[10];(2)由于转录因子也是蛋白质,因此,随着细胞总蛋白质合成速率的下降,大多数转录因子的合成速率也会跟着下降,从而导致总蛋白质合成速率的下降。
肿瘤抑制蛋白P53与组蛋白去乙酰化酶(HDAC2)转录水平呈明显的正相关性。利用Pathcards数据库(http://pathcards.genecards.org/)中的“p53 Signaling SuperPath”发现,大量的HDAC家族蛋白均参与了P53相关的超级通路,提示,随着细胞衰老导致的P53水平的逐渐上调,组蛋白H3和H4整体乙酰化水平也会逐渐下调,使DNA的转录速率也随着逐渐下调,从而下调了总蛋白质的合成速率。
3 决定细胞衰老的部位是细胞核
1958年,Yoshida将密伊乐藻(Elodoa den.se)幼叶浸在0.2mol/L.CaCl溶液中,使叶肉细胞发生质壁分离,一些细胞的原生质体分为两部分,一半有核,一半无核,在显微镜下观察培养8小时,发现处于有核原生质体中的叶绿体,经历了衰老和结构破坏的过程,而无核原生质体的叶绿体仍保持绿色和连续积累淀粉[11];1965年,Wright和 Hayflick把年轻的细胞核植入去核的衰老的细胞质中,结果细胞恢复了年轻,并按年轻的细胞核该有的分裂次数继续分裂下下去。相反,把衰老的细胞核植入去核的年轻的细胞质中,结果细胞出现衰老表型,细胞分裂次数大为减少。
以上两个实验说明决定细胞衰老的部位是细胞核,因此,细胞质中各种衰老性变化都不是导致细胞衰老的原因,而是由细胞核衰老介导的结果。也就是说,细胞质中的线粒体功能障碍只是细胞核衰老介导的结果,年轻的细胞核能够让功能障碍的线粒体恢复正常。反过来,衰老的细胞核也能够让年轻的线粒体产生功能障碍。
4 细胞衰老是由端粒DNA和核糖体DNA共同调控的
端粒DNA和核糖体DNA(rDNA)都是属于多拷贝的串联重复DNA阵列,很不稳定,很容易因为频繁的转录等因素导致拷贝丢失。几十年来就有人认为,端粒DNA和核糖体DNA(rDNA)与衰老有关,但两者对衰老的机制都没有统一在一起,也不清楚端粒DNA阵列和rDNA阵列缩短到底是导致细胞衰老的原因,或仅仅是细胞衰老的结果。而且很多研究都认为,过短的端粒或过短的rDNA阵列,是通过触发基因组不稳定性和DNA损伤来诱导细胞衰老的,很明显这种解释是错误的,因为体内衰老细胞的端粒和rDNA阵列的剩余长度,还不至于发生基因组不稳定性,以及为什么细胞是逐渐由年轻走向衰老,而不是突然衰老的?
很多证据表明,端粒并不是限制细胞传代次数和衰老的唯一因素,因为人类有很多细胞端粒不缩短,但传代次数仍然是有限的[12],以及用端粒酶维持较长的端粒,仍然无法阻止复制性衰老[13-14]。此外, 酵母菌、 线虫和果蝇的细胞的端粒也不会缩短。有些鸟类甚至年龄越大端粒越长[15]。因此,除了端粒,应该还有另一种类似端粒的东西共同调控着细胞的衰老。“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”认为[16],导致细胞衰老的根本原因,是由细胞核中的端粒DNA和核糖体DNA(rDNA)通过P53介导的。
各种衰老细胞都会上调P53,P53/P21/P16信号通路在调节衰老方面发挥着重要作用[17]。 约有一半的肿瘤细胞p53基因突变,P53基因缺失可以使细胞无限增殖[18],说明P53是调控细胞衰老的主控因子。因此,各种与衰老有关的标志或表型,可能都是由P53直接或间接介导的。
通过敲低小鼠和人类原代细胞中的 45S rDNA拷贝数,结果:衰老标志物P53、P21、P16和SA-β-GAL都显著上调,端粒长度、细胞活力和细胞传代次数都显著减少。此外,还检测了小鼠的衰老细胞和hESC与hiPSC,发现衰老细胞的端粒长度和45S rDNA拷贝数都显著减少了,hESC与hiPSC的端粒长度和45S rDNA拷贝数都显著增加了,这些数据有力的证明了细胞衰老的根本原因是由端粒和 45S rDNA 共同调控的,而且rDNA对衰老的权重大于端粒(https://www.biorxiv.org/content/10.1101/2024.07.23.604700v1)。
人类45S rDNA 位于近端着丝粒染色体13号,14号,15号,21号,22号的短臂和卫星体之间。近端着丝粒染色体指的是有些染色体的着丝粒在末端[19]。P53分布在端粒和rDNA等部位[20-21],并通过降解机制保持P53的收支平衡。相比于衰老细胞,年轻细胞的端粒和rDNA阵列较长,能快速降解P53,使P53保持低水平。因此,随着细胞中的端粒DNA和rDNA的的阵列逐渐缩短,就会导致P53水平逐渐上调,从而使细胞从年轻状态经历到衰老状态。
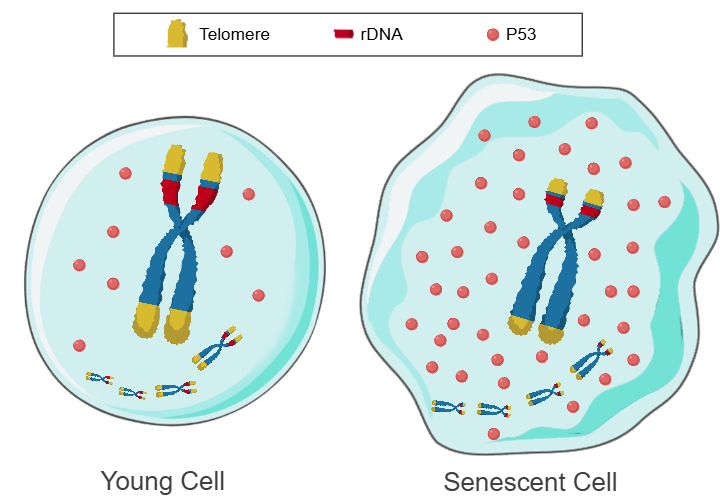
图1:Telomere DNA and ribosomal DNA co-regulation model for cell senescence
a.染色体上长阵列的端粒和rDNA,P53迅速降解,P53水平低,细胞年轻。b.染色体上短阵列的端粒和rDNA,P53缓慢降解,P53水平高,细胞衰老。
5 rDNA也调控非分裂性细胞的衰老
虽然大多数分裂性细胞的衰老是由端粒和rDNA共同调控的,但是,哺乳动物的一些终末分化细胞、蠕虫和果蝇的体细胞,染色体DNA通常不复制,端粒就不易缩短。为了使终末细胞也会逐渐衰老,必须还要有另一套能让细胞P53水平上调的装置,例如,果蝇在40天寿命里,rDNA阵列会缩短一半[22]。从理论上讲,如果rDNA只是为了合成rRNA,那么,rDNA可以多拷贝但无需串联重复。然而,鉴于rDNA具有高度不稳定性、多拷贝的串联重复,能与长寿蛋白Sir2和衰老总控蛋白P53结合,以及rDNA转录时很容易导致拷贝丢失来看,rDNA的结构和属性是专门为了作为第二套控制细胞分裂和衰老的“计数器”而设计的。
6 基因如何实现程序化表达
发育与衰老的过程是基因的程序化表达的过程,但是,在年轻的细胞和衰老的细胞中,染色体上的各种基因的排列顺序和拷贝数通常是不会随着时间的推移而改变的,那么,是什么力量在驱动固定不变的基因沿着时间轴进行程序化表达(顺序表达)的?
大约1/10的人类基因启动子含有P53结合位点,因此可被归类为P53反应基因[23]。P53不但能够下调总蛋白质合成速率,而且又是一种能同时沉默和激活众多基因的转录因子。因此,只要P53的水平逐渐上调,总蛋白质合成速率就会逐渐下调,一部分基因会特异性的上调和下调,从而让细胞在功能下降的同时,基因也呈现出程序化的表达。由于不同分化类型的细胞有着不同编程的遗传程序,因此,在衰老过程有着不同的基因表达模式,例如,在衰老的造血干细胞,大约有1500个基因表达随着年龄的增长而发生显著的上调和下调[24]。因此,端粒和rDNA相当于索引,基因组相当于数据库,在同一分化类型的细胞中,不同长度的端粒和rDNA阵列就会有不同的基因表达谱。例如,随着端粒逐渐缩短,DUX4表达活性最多上调10倍[25]。端粒能通过改变表观遗传影响基因表达[26]。不同长度的rDNA阵列会有不同分布的异染色质/常染色质,从而改变了基因表达谱[27-28]。P53也可以通过直接或间接影响DNA (cytosine-5)-甲基转移酶3A(DNMTs),例如,P53会结合到DNMT1基因的启动子上[29],因此,随着P53水平的增龄性变化,就会改变表观遗传模式和基因表达谱。DNMT3A水平会随增龄而下调,而该基因缺失的造血干细胞会在不同的染色体区域表现出甲基化升高或者降低的现象,从而改变了基因表达谱[30]。
程序的要点是时钟、指令、序列、驱动机制和运行结果,据此推测,基因程序的运行机制是这样的:随着端粒DNA阵列和/或rDNA阵列(时钟)的缩短,P53(一级指令)就会沿着时间轴产生“浓度梯度,由于P53会与多种基因的启动子和增强子(序列)结合,影响包括各种转录因子(二级指令)基因在内的多种基因表达(驱动机制),从而使有些基因表达上调,有些基因表达下调(运行结果)。
7 端粒DNA和rDNA是驱动遗传程序运行的计时器的最佳候选者
虽然很多基因会影响细胞衰老速率和衰老标志物水平,但并不是决定衰老的根本原因。总的来说,要让不变的基因实现程序化表达,需要一个计时的装置来驱动。而多拷贝的串联重复序列的端粒DNA和rDNA阵列完全具备计时器的各种要素:(1)细胞分裂时它能复制一份,以让每个子细胞都有一份;(2)在体细胞中缩短的那部分阵列长度,会在生殖细胞中得到补充,而且储量在个体的所有细胞中须最多,以满足从胚胎发育到成熟衰老的整个生命周期的消耗[31,22];(3)阵列缩短速率具有延展性,以适应不同物种和同一物种的不同类型细胞的不同发育与衰老速率的需求;(4)在体细胞中,只能单向消耗端粒DNA或/和rDNA,以让P53能沿着时间轴生成“浓度梯度”,形成“时间量度”,使基因进行程序化表达,调控个体的生长发育、成熟衰老和死亡;(5)各种影响衰老速率的外部因素或基因本身,都是通过影响阵列的缩短速率来实现的;(6)由于个体的寿命可以达到百年以上,例如,格陵兰鲨鱼寿命可达400年,因此,候选为驱动遗传程序运行的驱动物质(相当于沙漏计时器中的沙子)必须非常稳定,没有半衰期。而蛋白质、RNA、线粒体DNA(mtDNA)、以及DNA和组蛋白的化学修饰,这些都是很不稳定,有半衰期,都处于不断降解和补充的动态平衡中,例如,DNA甲基化与去甲基化,组蛋白的乙酰化与去乙酰化,都是同时进行的,因此无法形成时间量度,不具备计时的属性。也就是说,衰老根本原因不在于RNA、蛋白质、mtDNA和表观遗传修饰上,往这些方向研究永远找不到衰老的根本原因。
一个理论是否正确,要看这个理论是否具备自洽性,既然端粒DNA和rDNA阵列是驱动基因群程序化表达的驱动物质,那么,在体细胞中缩短的端粒DNA和rDNA[32],在生殖细胞或胚胎的早期细胞中必须要能够补充,否则,生命无法进行世代轮回。或者说,端粒DNA和rDNA的阵列就象”沙漏计时器”中的沙子,沙子漏完后需要再次补充。幸运的是,已有证据表明,在体细胞中缩短的端粒DNA和rDNA阵列,可以在胚胎早期细胞或生殖细胞中得到补充[31,22]。检测也发现,hESC和hiPSC的返老还童机制不是因为表观遗传重编程,而是因为端粒DNA阵列和45S rDNA阵列的长度都显著增加了(https://www.biorxiv.org/content/10.1101/2024.07.23.604700v1),因此,“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”是唯一高度可信的细胞衰老理论。
8 为什么用端粒时钟衡量生理年龄不如表观遗传时钟准确
随着年龄的增长,全基因组DNA甲基化水平会逐渐的下降,据此可以人为赋予一种用来测量衰老的表观遗传时钟[33]。论准确度,表观遗传时钟比端粒时钟更精准。至于为什么用端粒时钟衡量生理年龄不如表观遗传时钟准确,可能有如下3个原因:
(1)端粒能调控表观遗传[26],但从端粒到表观遗传的流程较长,途中会受到各种因素的影响;(2)人类的睾丸、卵巢、小脑、阴道、骨骼肌、和甲状腺等组织的端粒长度随年龄增长缩短不明显[12]。成熟果蝇的体细胞端粒也不会缩短。体重只有7克的布氏鼠耳蝠(Myotis brandti),寿命长达40年左右,端粒也不会缩短[34];(3)虽然已发现端粒和rDNA阵列的缩短处于同一水平[32]。端粒缩短会导致rDNA阵列不稳定性,而rDNA阵列不稳定会促使rDNA阵列缩短,这可解释为什么端粒和rDNA阵列的缩短处于同一水平。例如,人类细胞中的端粒破坏被证明会立即导致大规模的核仁缺陷,随后是rDNA阵列的不稳定性[35],但不总是这样,端粒缩短速率一开始很快,然后慢慢减速[36-37],最后开始反弹,例如,到了80~90岁,男性和女性的端粒长度略有增加[38-39]。老年裸鼹鼠端粒反而变长[40],出现这种反常现象可能是端粒对rDNA阵列缩短进行代偿。对衰老细胞进行rDNA拷贝数敲除也证明了这一点,rDNA阵列缩短会导致白细胞端粒长度的增加(https://www.biorxiv.org/content/10.1101/2024.07.23.604700v1)。
9 个体衰老是由成体干细胞的“复制性衰老”导致的
细胞外基质和细胞内的交联变性或错误折叠的蛋白质都是可以降解更新的,衰老组织之所以会积累这些垃圾,是因为遗传程序关闭了防御机制,因此,个体的衰老不可能是大分子垃圾积累导致的。
除了心脏,所有器官组织中都发现了驻留的干细胞,这些干细胞通过自我复制和细胞分化,持续产生新的干细胞和“功能细胞”,维持着组织和器官的稳态与修复。但是,干细胞和功能细都会因为细胞衰老、基因突变或病毒感染等因素而被免疫系统清除掉,为了补充这些损耗掉的细胞,干细胞要经历反复的有丝分裂,由于成体干细胞的分裂次数是有限的,而且每分裂一次就会比上一代更老一些,从而产生了复制性衰老。而由衰老的成本干细胞分化的功能细胞也是衰老的功能细胞,从而导致组织、器官和个体的衰老,因此,导致个体衰老的根本原因,归根结底是由成体干细胞本身的复制性衰老导致的。
如果成体干细胞不会发生复制性衰老。那么,由DNA损伤、细胞毒性化合物和癌基诱导等等因素导致的细胞衰老,并由此损耗掉的成体干细胞和功能细胞,都可以由成体干细胞通过自我复制和细胞分化补充上去,也就是说,如果成体干细胞不会发生复制性衰老,个体就能青春永驻。
如上所述,复制性衰老是由端粒和rDNA阵列缩短导致的,例如,小鼠造血干细胞因为mTOR1激活导致rDNA阵列缩短[41],抗衰老药雷帕霉素能通过抑制mTOR1来抑制细胞复制,减缓细胞复制性衰老。
1998年,我在“YANJING MEDICAL CORRESPONDENCE”上发表题为《衰老的机理意义及治疗》的专著指出,导致个体衰老的根本原因,是组织中的干细胞的端粒等重复DNA阵列缩短导致的,而不是干细胞数量的减少,并且还排除了细胞核DNA和mtDNA突变等看似导致细胞衰老的因素。电离辐射会极大的导致细胞核DNA和mtDNA突变,然而,电离辐射不但不会缩短小鼠寿命,反而延长了寿命,这个重要发现也可以排除衰老是由DNA突变导致的假设[42]。因为生物在漫长的演化过程中,已经产生了冗余的克服这些问题的解决方案,而遗传程序会逐渐关闭这些解决方案。例如,mtDNA突变可以通过“线粒体自噬”等途径来解决[43],因此,突变的mtDNA的积累仅仅是衰老的结果。衰老的细胞核之所以会积累突变的DNA,也是因为逐渐关闭了DNA修复系统和清除衰老细胞和突变细胞的免疫系统。因此,个体衰老的细胞核DNA和mtDNA的突变积累理论,无法解释为什么寿命从不到20天的秀丽隐杆线虫,到寿命超过400年的格陵兰鲨鱼的寿命为何有着如此巨大的差距。
关于个体衰老与干细胞关系的论证,2007年,英国的Anastasia 等人在《Nature》杂志发表的论文指出:干细胞对人体自我修复和组织再生至关重要,干细胞数量的减少是导致人体衰老的主要原因[44]。Anastasia等人的结论是错误的,首先,人体干细胞不是不够用,而是存在着干细胞数量的控制系统,例如,在秃头患者的头皮中,毛囊干细胞还在,只是无力长毛发[45]。老年小鼠造血干细胞数量增加5倍[46]。另外,人体存在可清除DNA损伤的免疫系统,因此,免疫系统衰老才是导致DNA突变的细胞在组织中积累的罪魁祸首,而免疫系统的衰老是造血干细胞衰老衍生的结果,因为由衰老的造血干细胞分化补充的各种免疫细胞也是衰老的免疫细胞;2010年,美国的Sahin和Depinho也在《Nature》杂志上发表论文指出,导致个体衰老的主要原因是组织中的干细胞的端粒缩短和mtDNA突变造成的[47]。Sahin和Depinho的结论也是错误的,首先,突变的mtDNA会通过称为“线粒体自噬”等途径选择性的清除掉[43],其次,线粒体障碍的细胞无法与健康细胞竞争而被淘汰。并且已发现,增加mtDNA突变的小鼠模型没有任何加速衰老的迹象,也不会缩短寿命。超氧化物歧化酶的杂合突变虽然导致氧化损伤和mtDNA突变增加,但并没有缩短小鼠的寿命。
理论上,用年轻的成体干细胞替换衰老组织中衰老的成体干细胞,就能让衰老的组织返老还童。但是,移植异体的干细胞会被免疫清除掉,移植经过扩增的自体干细胞,由于扩增时会产生复制性衰老,移植来自于自体细胞的诱导性多能干细胞(iPSC)分化的成体干细胞,由于可检测的DNA损伤超过70%[48],也会被免疫清除掉[49],因此这些方法都行不通。唯一可行的办法是增加自体的成体干细胞中的端粒和rDNA 阵列的长度。
一般认为人体有78个器官,但组织学家认为每块骨头都是一个器官(骨骼干细胞也有多种亚型[50]),因此,人体总共有284个器官。再加上头发、眉毛、睫毛、鼻毛、腋毛、阴毛和体毛的毛囊干细胞也不是完全相同的干细胞,因此,包括各种成体干细胞亚型,人体中可能有超过300种成体干细胞。因此,只移植几种年轻的成体干细胞是无法让衰老的个体完全返老还童。
二 12种衰老标志的因果关系
很多人都认为,弄清衰老的因果关系可能要花几十年[51],由于P53是衰老的主控因子,接下来我们将对衰老的十二大标志[2]与P53联系起来进行因果关系的论证:
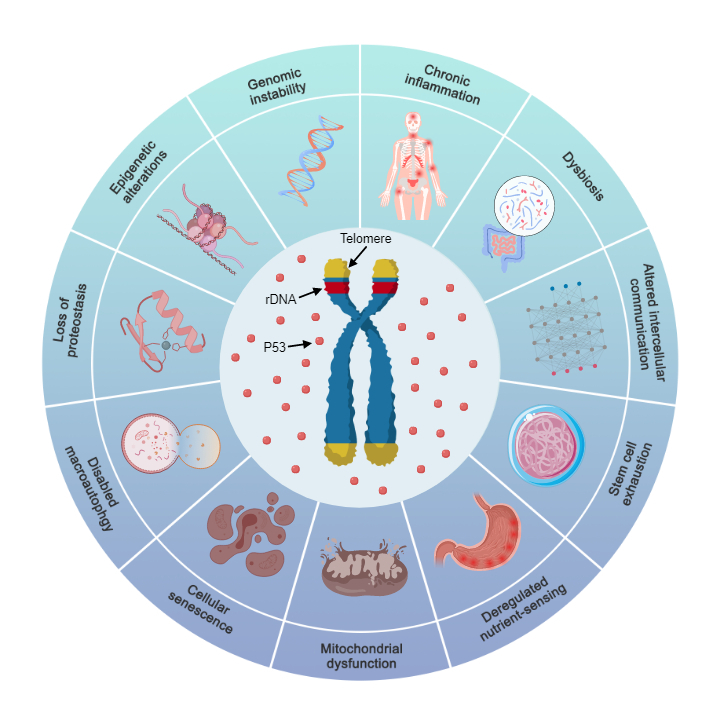
图2:端粒和rDNA通过P53介导的衰老十一大标志
1 基因组不稳定(genomic instability):随着年龄的增长,基因组会变的越来越不稳定,使染色体更容易发生畸变,DNA更容易发生突变,以及本该沉默的重复元件内源性逆转录病毒(Endogenous retroviruses, ERVs)的激活。
那么,是什么原因导致基因组不稳定性的?
端粒或rDNA阵列太短会导致基因组不稳定性和触发DNA损伤反应[52]。然而,小鼠和人类的初始端粒长度分别为50kb和15kb,小鼠端粒缩短率为每年6420 bp,相当于人类端粒缩短速度的100倍[53],因此,老年小鼠的剩余端粒比人类初始端粒还长,还不至于发生基因组不稳定性。实际上,人类衰老细胞的端粒和rDNA还剩初始的一半长度,因此,衰老细胞发生的基因组不稳定性与端粒和rDNA阵列的物理长度无关。
异染色质区域的DNA是处于超甲基化和组蛋白低乙酰化的状态,这样组蛋白才能紧密的包裹住DNA,使基因组保持稳定性。而DNA甲基转移酶DNMT1对于保持异染色质DNA超甲基化状态起着重要的作用。
P53会结合到dnmt1基因的启动子上[29],因此,随着端粒和rDNA的阵列逐渐缩短,不断上调的P53会导致DNMT1基因的表达量逐渐下调,降低了异染色质区域DNA的甲基化水平,导致“异染色质丢失“和基因组不稳定性。
基因组不稳定性与着丝粒的功能失活有关,是导致染色体分裂错误的重要原因之一。而着丝粒的功能依赖于一种特异性组蛋白变体CENP-A。CENP-A在衰老细胞中以P53依赖的方式下调[54]。
SIRTs会与端粒和rDNA结合,保持端粒和rDNA区域的稳定。而P53会抑制七个SIRTs蛋白表达,从而导致端粒和核仁区rDNA不稳定。在酵母菌,Sir2结合在rDNA区域[55],Sir2与Sir3和Sir4结合在端粒[56-57]。哺乳动物中SIRT7定位在核仁,保持rDNA稳定[58]。
异染色质丢失会让沉默的ERVs转录,再加上P53也能够激活ERVs转录[59],产生了无菌性的慢性炎症。慢性炎症被认为会促进衰老[60],然而,用逆转录酶抑制剂(NRTIs)还没观察到能延长野生型小鼠的寿命[61],说明ERVs的释放只是衰老的结果。
异染色质一般包含高密度的重复DNA元件,如端粒序列、卫星序列、rDNA重复序列和转座元件,因此,异染色质主要位于着丝粒、端粒和核仁区域[62]。如上所述,端粒DNA和rDNA都是属于多拷贝的串联重复阵列,很不稳定,很容易因为频繁的转录等因素导致拷贝丢失,因此,增加基因组的稳定性能够减缓端粒和rDNA的阵列的缩短[16,26]。
根据“Telomere DNA and ribosomal DNA co-regulation model for cell senescence”[16]推测,不同物种寿命的差异取决于端粒和rDNA的阵列的缩短速率。已观察到,端粒和核仁区的rDNA的阵列的缩短速率处于同一水平[32]。热量限制(CR)和雷帕霉素就是通过抑制mTOR1来抑制核仁区的45S rDNA转录,从而减少45S rDNA拷贝丢失来延缓衰老的。以下几个例子也可佐证抑制45S rDNA转录能延缓衰老:
抗衰老药亚精胺可显著下调细胞内mTORC1的活性,从而抑制rDNA转录[63];RPL22可以通过破坏核仁区域异染色质结构,促进了45S rDNA转录并引发细胞衰老[64];4EBP1基因敲除小鼠激活了mTORC1,促进45S rDNA转录,使小鼠的心脏衰老加速[65];少吃能长寿是有依据的,过量进食的果蝇会过度刺激mTOR1,从而加速45S rDNA阵列缩短[66];小鼠造血干细胞中的45S rDNA阵列会随着mTOR1的激活而缩短[41];IL-11会上调ERK-AMPK-mTORC1轴,随着小鼠年龄的增长会上调IL-11,用抗体抑制IL-11能够延长小鼠寿命[67]。
和年轻小鼠相比,老年小鼠会积累更多的DNA突变,DNA突变不是导致细胞衰老的原因,而是免疫系统衰老的结果,因为衰老的免疫系统无法有效的监视和清除掉突变的细胞。研究也发现,DNA突变不会导致细胞衰老[68];鳉鱼能在不足6个月里大量积累DNA突变的细胞,并患上癌症[69],也说明了DNA突变细胞的积累是因为免疫系统的衰老。由于DNA损伤积累只是免疫系统衰老的结果,因此就无法解释寿命只有10几天的秀丽隐杆线虫到400多年的格陵兰鲨之间的寿命为何有着如此巨大的差距,也无法解释人体内一些寿命只有几天的细胞和长寿的心肌细胞之间的寿命为何有着如此巨大的差距。
肿瘤细胞基因组具有不稳定性,但肿瘤细胞能够永生,以及表观遗传年龄也不受辐射诱导的DNA断裂引起的基因组不稳定性的影响[70],说明细胞衰老不是由基因组不稳定性导致的。
2 端粒损耗(telomere attrition):端粒DNA阵列的长度会因为染色体DNA复制而缩短,是导致细胞复制性衰老的根本原因之一,因为延长端粒能显著增加细胞分裂次数和降低衰老标志物[71]、下调DNA损伤和癌症发生率[72]、使小鼠中位数寿命延长40%,体毛变黑[73]。但是,除了端粒之外,rDNA也是调控细胞衰老的主要原因[16],而且根据我们的检测,rDNA对衰老作用的权重大于端粒。因此,端粒缩短不仅是衰老标志,在因果关系中还是衰老原因。
端粒长度与DUX4基因表达呈负相关[25]。DUX4与MHC-I基因表达呈负相关,因此,随着端粒的不断缩短,DUX4表达会逐渐上调,MHC-I表达会逐渐下调,因此,DUX4上调会影响MHC呈递抗原[74],这就是衰老组织中积累衰老细胞和中老年人肿瘤发生率比年轻人高的原因之一。此外,端粒缩短增加癌症易感性的真正原因不是染色体不稳定性,而是较短的端粒会导致T细胞免疫缺陷[75]。在德系犹太百岁老人中,hTERT和hTERC突变拥有更长的端粒,更长的寿命和预防年龄相关疾病[76]。端粒最短组癌症发病率是端粒最长组的3倍,端粒最短组的癌症患者死亡率是端粒最长组的11倍[77]。
总的来说,增加端粒长度会让免疫系统更年轻,从而减少组织中衰老细胞的积累和肿瘤的发生率。
3 表观遗传改变(epigenetic alterations):随着年龄的增长,DNA甲基化模式、组蛋白修饰等表观遗传特征会发生改变,影响了基因表达和细胞功能。
但是,表观遗传的增龄性改变也是由端粒和rDNA调控的[26-28]。P53会结合到dnmt1基因的启动子上介导表观遗传[29],因此,表观遗传改变不是驱动衰老的原因,而是端粒和rDNA阵列缩短产生的结果,因此,单纯干预表观遗传的抗衰老途径是行不通的。例如,在12个月大的野生型小鼠体内进行部分重编程(表达Oct4, Sox2, Klf4 和C-myc )并没延长寿命[78];部分重编程的野生型小鼠中位寿命仅提高了12% ,还不如小分子抗衰老药[79],增加这点寿命的原因可能与重编程用的山中因子Oct4、Sox2、Klf4和多西环素具有促进细胞再生有关。例如,促进血管生成能显著延长小鼠寿命[80];部分重编程结束后,发现端粒没有延长或略有缩短,衰老标志又开始积累,表观遗传年龄恢复到重编程之前的状态[81]。
生长激素能逆转DNA甲基化时钟[82],但生长激素会缩短小鼠寿命;Rudman对老年个体生长激素补充实验发现,生长激素可使60-80岁老人肌肉增加皮肤变厚,年轻10-20岁,但跟踪调查发现,长期补充生长激素的老人,寿命不但没有延长,反而衰老加速了;一位60岁的老人接受了生长激素释放激素(GHRH)基因治疗后,生理年龄出现逆转,PhenoAge时钟年龄下调了28.6岁,表观遗传年龄下调了6岁,但端粒年龄却比同龄人老了7个月[83]。
生长激素下调表观遗传年龄和缩短寿命的悖论,其实就是促进蛋白质和ATP合成,检测时显示年轻状态,实际上是加速细胞分裂,透支了有限的端粒和rDNA阵列,或者说是透支了有限的细胞分裂次数,就像蜡烛火焰越大烧的越快,很多抗衰老措施其实也是透支有限的细胞复制次数,抗衰不成反而折寿。
4 蛋白质稳态丧失( loss of proteostasis):随着年龄的增长,组织细胞会积累错误折叠和错误翻译的蛋白质,从而影响了细胞的功能。
热休克蛋白HSP70的基因表达水平下降是导致蛋白质稳态的丧失的主要原因,65岁和85岁年龄组的单核细胞HSP70表达水平明显低于25岁年龄组[ 84]。
美国国家老龄研究所最严格的的衰老干预测试:ITP(http://www.nia.nih.gov/research/dab/interventions-testing-program-itp)在遗传异质性小鼠模型(UM-HET3)中测试了多种化合物对寿命的影响。替普瑞酮(GGA)对哺乳动物组织中的热休克蛋白表达有诱导作用,保证蛋白质能正确折叠,但ITP测试发现GGA并不能延长小鼠寿命[85],说明蛋白质稳态的增龄性丧失不是导致细胞衰老的原因,而是细胞衰老的结果。
P53能够结合到SIRT1基因的启动子上,从而抑制SIRT1的转录[86]。SIRT1会促进热休克蛋白HSP70的转录[87]。因此,端粒和rDNA的阵列的缩短使P53水平上调会抑制SIRT1基因表达,从而下调HSP70水平,增加了错误折叠的蛋白质。
5 大自噬失能(disabled macroautophagy):自噬的任务是清除异常的蛋白质和细胞器,以此保持细胞健康,随着年龄增长,这一机制变得不那么高效。
敲除或药物抑制P53可以在人类、小鼠和线虫细胞中诱导自噬[88]。研究人员对自噬促进长寿的“共识”提出了质疑[89]:来自蠕虫和哺乳动物的研究数据显示,线粒体的通透性决定了自噬对衰老的影响,在线粒体通透性增强的情况下,增强自噬反而有害健康;小鼠卵巢衰老与颗粒细胞自噬增强有关[90];氯喹(CQ,Chloroquine)能抑制自噬。用氯喹治疗野生型衰老的大鼠五个月,大鼠中位寿命延长了6%,最大寿命延长了13%,这与有史以来在小鼠模型中测试的一些最好的抗衰老化合物不相上下[91];增强自噬能促进年轻蠕虫的健康,但也能推动衰老进程,关闭老年蠕虫自噬,神经元和身体健康也有了显著改善,寿命显著的延长了50%[92]。因此,亚精胺和雷帕霉素能增强自噬的抗衰老药不是靠增强自噬来延长寿命的,而是靠通过抑制mTOR1来下调rDNA转录,以此减少rDNA陈列缩短来延长寿命的。
由于提高自噬对寿命的干预往往自相矛盾,说明自噬功能障碍不是衰老的原因,而是衰老的结果。
6 营养感应失调(deregulated nutrient-sensing):衰老和代谢疾病的产生与营养感应(例如AMPK和SIRT1)的调节息息相关[93],营养感应能力会随着衰老而下调。
如上所述,端粒和rDNA的阵列缩短会导致P53水平上调,而P53上调又会导致总蛋白质合成速率下调和一部分基因特异性的上调或下调,因此,各种与营养感应有关的受体与配体蛋白合的成速率的下调和改变,是导致营养感应随着年龄的增长而下调的主要原因。因此,超长端粒小鼠对胰岛素和葡萄糖的耐受性更强、更低的胆固醇和低密度脂蛋白和更瘦的体型[72]。
过氧化物酶体增殖物激活受体-γ共激活因子-1α(PGC-1α)是一个关键的转录因子,在葡萄糖稳态中发挥作用,对能量平衡、糖尿病基本途径有很大的影响,在衰老过程中,PGC-1α水平下降从而导致健康问题[94]。SIRT1通过对PGC-1α的去乙酰化作用,可以阻止蛋白酶体降解PGC-1α。而P53会通过抑制SIRT1表达来促进PGC-1α降解,因此,老年小鼠在空腹下无法维持血浆葡萄糖水平[95]。
7 线粒体功能障碍(mitochondrial dysfunction):线粒体是细胞的能量工厂,随着年龄的增长,ATP产量会逐渐下降,影响细胞的能量供应和健康状态。
蠕虫添加抗氧化剂维生素C,线粒体ATP产量增加了2.5倍以上,在治疗后的第8天,老年色素-脂褐素(Lipfuscin)含量增加了约18%,寿命明显缩短了。相反,多西环素能抑制ATP的产生,年轻的蠕虫在浓度为13μM(6μg/ml)的多西环素下,寿命延长了72.8%。在处理后的13天,脂褐素的含量下降了约50%[96]。多西环素也能延长早衰小鼠寿命[97]。
短端粒会导致线粒体功能障碍[98]。50个编码线粒体蛋白的核基因中有25个基因的表达与rDNA剂量呈显著正相关[99]。超长端粒的小鼠模型线粒体功能也更好、较少的DNA损伤[72]。如上所述,P53会抑制PGC-1α和PGC-1β的表达。PGC-1α/β表达的下调反过来会导致线粒功能障碍,下调ATP产量。
小鼠线粒体超氧化物歧化酶(SOD2)的杂合突变虽然导致氧化损伤和线粒体DNA(mtDNA)突变增加,但并没有缩短寿命[100]。和野生型小鼠相比,mtDNA突变多50倍的小鼠,没有骨质疏松、秃顶或者生育力下降等过早衰老的迹象[101]。至今没有发现通过增强线粒体来有效延长寿命的报道,说明线粒体功能障碍只是衰老的结果。
8 细胞衰老(cellular senescence):细胞衰老会导致组织、器官和个体的衰老,衰老细胞是不能再分裂但会分泌炎症因子的细胞,衰老细胞积累起来会影响组织的更新和修复。
2011年,Baker DJ等人让野生型老年小鼠服用可以清除衰老细胞的药物,虽然健康得到了改善,但寿命没有延长[102]。从青年时期开始服用清除衰老细胞的达沙替尼和槲皮素组的雌性小鼠却加速了衰老[103]。用达沙替尼和槲皮素清除人类衰老细胞,观察到表观遗传年龄显著增加,同时端粒长度显著缩短[104]。清除衰老细胞的ABT-263 治疗加速老年雌性小鼠卵巢衰老[105]。这是因为清除衰老细胞会刺激周围年轻细胞复制和分化,以此来填充清除掉衰老细胞留下的空穴,从而产生复制性衰老。
9 干细胞耗竭(stem cell exhaustion):除了心脏,所有器官组织中都发现了驻留的成体干细胞,负责组织细胞的更新与修复。因此,成干细胞需要反复的分裂。但是,成体干细胞复制次数是有限的,而且每复制一次就会比上一代更老一些,从而产生复制性衰老。好比NK细胞的数量虽然会发生增龄性的增加,但NK细胞清除肿瘤细胞的效率反而下降了很多,这是老年人更容易患上癌症的重要原因之一[106]。在秃头患者的头皮中,毛囊干细胞还在,只是无力长毛发[45]。老年小鼠造血干细胞数量增加5倍[46]。
总的来说,干细胞耗竭与干细胞数量减少无关,而与干细胞本身的复制性衰老有关。个体组织中驻留的成体干细胞因为反复的分裂,端粒和rDNA阵列会不断缩短,从而导致P53逐渐上调,产生了复制性衰老,因此,成体干细胞中的端粒和rDNA阵列缩短是导致个体衰老的根本原因。
10 细胞间通讯改变(altered intercellular communication):细胞间通信是维持组织功能的关键,衰老过程中,这种通讯会受到干扰。
细胞衰老过程是基因表达谱逐渐改变的过程[107],不同年龄有着不同的基因表达谱,因此,各种通讯分子的受体与配体的数量和活性也会随着年龄的增长而改变,再加上衰老细胞产生的炎症信号的干扰,结果导致了细胞间通讯改变。
11 慢性炎症(chronic inflammation):衰老伴随着慢性低度炎症的增加,这种慢性炎症状态与许多与年龄相关的疾病有关。
衰老的细胞核和线粒体会释放DNA碎片到细胞质,机体以为被病毒感染了,产生了无菌性的慢性炎症,以召唤免疫细胞过来清除掉衰老细胞[60,108]。
衰老的细胞会产生一组炎症因子或称为衰老相关分泌表型(Senescence-associated secretory phenotype,SASP),而P53会激活下游基因p21,然后P21会通过降低Rb的磷酸化促进SASP的产生[109]。P53会结合在炎症因子IL-22基因的启动子促进IL-22基因转录[110]。阿尔茨海默病(AD)脑组织处于炎症状态,AD大脑皮质神经元和皮肤有着高水平的P53[111-113]。P53能诱导理应被沉默的重复元件ERVs的表达引发炎症[59]。
作为炎症因子IL-11会随小鼠年龄增加而上调,激活ERK-AMPK-mTORC1信号通路。用抗体抑制 IL-11 ,可使小鼠平均寿命延长 22%以上[67]。由于IL-11 会上调mTOR1信号通路,因此,抑制IL-11 延长小鼠寿命的机制可能不是因为降低了炎症水平,而是相当于吃mTORC1抑制剂的抗衰老药雷帕霉素。因为巨噬细胞迁移抑制因子拮抗剂(MIF098)也能够抑制机体内的慢性炎症。能增加NAD+的抗衰老药NR也能减少52.6%炎症因子的释放[114],但经ITP测试发现:NR反而使小鼠寿命缩短3%。MIF098对寿命没影响[115]。ERVs会引发炎症,但没发现逆转录酶抑制剂(NRTIs)能够延长野生型小鼠寿命[61]。与短命的鲨鱼物种相比,参与激活NF-kB信号通路的三个基因家族(TNF、TLR、LRRFIP)的拷贝数在格陵兰鲨中显著增加。作者认为,炎症基因显著增加了,导致免疫力增加了,有助于格林兰鲨鱼极长寿[116]。说明慢性炎症只是衰老的结果,靠抑制炎症来延缓衰老的措施宣布失败。
P53会抑制七个SIRTs蛋白表达,而SIRT3缺乏加剧了P53/Parkin介导的线粒体自噬抑制[117],P53抑制Parkin介导的线粒体自噬并促进小鼠心脏线粒体功能障碍[118], 当线粒体自噬受损时,mtDNA会从线粒体泄漏到细胞质中,激活cGAS-STING通路,引发炎症反应[119]。因此,P53水平的增加会抑制线粒体自噬,使mtDNA泄漏到细胞质引发无菌性炎症。
12 生态失调(dysbiosis):这是指身体内部环境的失衡,包括微生物群落的变化,这会影响我们的健康。
给老年鳉鱼移植年轻鳉鱼的肠道菌群,能够延长老年鳉鱼的寿命[120],但是,喂年轻鳉鱼肠菌前要先用抗生素先杀死老年鳉鱼的肠菌,如上所述,部分重编程用的多西环素能延长蠕虫和小鼠寿命,因此,抗生素本身带来的有益作用也能延长老年鳉鱼的寿命,再说鳉鱼寿命很短,新建立的菌群还没完全破坏就快死了,因此,年轻粪便能延长老年鳉鱼的寿命,不一定能延长寿命较长的小鼠寿命,更别提延长人类寿命了;最近有一例自相矛盾和截然相反的发现:来自老年宿主的肠道菌群,在年轻小鼠中具有延长寿命的有益作用[121]。
通过菌群移植,将老年个体肠道菌群恢复至年轻组成,可能无法有效逆转衰老[122];长寿人群的微生物独特性可能与健康老龄化无关[123]。肠道微生物群落会随着年龄增长发生显著改变[124]。而肠道分泌物特别是胆汁酸是决定微生物群丰度,多样性和代谢活性的重要决定因素[125-126]。P53通过调控小异二聚体伴侣(SHP)来调节胆汁酸合成,进而影响胆汁酸稳态。P53通过其反应元件(P53REs)抑制胆汁酸合成酶CYP7A1和CYP8B1的表达[127]。P53可以显著上调参与胆汁酸羟化(如CYP3A11和CYP2B10)和磺化(如SULT2A1)的酶基因表达,同时促进胆汁酸外排转运蛋白(如ABCC2、ABCC3和ABCC4)的表达[128],因此,胆汁酸组成和水平会随着年龄的增长发生变化[129-130]。因此,在饮食习惯不变的情况下,肠道微生物群落组成的增龄性变化是由消化道的分泌物增龄性变化决定的,说明肠道菌群失调是个体衰老的结果,而不在于微生物的本身。
肠道黏膜层是肠道阻止细菌进入血液的屏障,作为保健品的AKK菌,就是嗜黏蛋白阿克曼菌,靠吃肠道黏膜层里的黏蛋白为生。给予正常野生型小鼠口服Akk菌,粪便里的啮齿类柠檬酸杆菌数量大幅度增加,啮齿类柠檬酸杆菌还突破肠道屏障,闯进了血液,引发系统性感染,并导致结肠组织出现了严重的病理损伤[131]。
俄国诺贝尔奖得主Илья Ильич Мечников(1845-1916)认为通过喝酸奶改变肠道菌群能够大幅度延长寿命,他自己晚年也天天喝酸奶,活了71岁。后来有人调查统计结果,喝酸奶的人并非比不喝酸奶的人更长寿。
综上所述,由P53介导的衰老11大标志参见(表1)。
结论
随着研究的持续下去,还会陆续出现新的衰老标志物,衰老标志又不是衰老原因,没完没了的,就象膨胀的宇宙。例如,又有一个新的衰老标志物被提出:免疫球蛋白IgG,IgG能直接引起人和小鼠的巨噬细胞及小胶质细胞衰老,并释放炎症因子[132],这是因为,无用的废物积累都会产生毒物兴奋效应,表现出衰老的症状。例如,微塑料积累会激活衰老标志物SA-β-半乳糖苷和NF-kB、IL6、TNF-a和CD68等关键炎症标志物是一样的[133]。最近一篇综述又把衰老标志从12个增加到14个[134]。
其实很多衰老理论和干预措施都是错误的,这篇预印本进行了专门批判[135]。总的来说,目前学界都在蛋白质和RNA层面来研究衰老机制,而我们通过逻辑推演和实验研究都认为,决定细胞衰老和复制次数的驱动物质是端粒DNA和rDNA。或者说,调控衰老的第一阶层是在DNA水平,表观遗传和蛋白质等等只是作为DNA水平调控的介导者。要想突破寿命的天花板,必须要延长端粒和rDNA阵列,除此之外,单纯逆转表观遗传或调节信号通路等等等等都无济于事。因此,在未来,对于各种干预衰老措施的金标准,必须要有端粒和rDNA的阵列长度的增加。对于小鼠的寿命对照实验,必须采用野生型小鼠,而不是早衰症小鼠。
综上所述,端粒和rDNA阵列缩短是导致细胞衰老的根本原因,因此,通过干预各种衰老标志物的抗衰老方法效果非常有限,更不可能逆转衰老,要想逆转衰老和大幅度延长寿命,最好的办法是增加组织中成体干细胞的端粒和rDNA的阵列长度。
参考文献
[1] de Magalhães JP. Distinguishing between driver and passenger mechanisms of aging. Nat Genet. 2024 Feb;56(2):204-211. doi: 10.1038/s41588-023-01627-0.
[2] López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023 Jan 19;186(2):243-278. doi: 10.1016/j.cell.2022.11.001.
[3] Sigal SH, Brill S, Fiorino AS, et al. The liver as a stem cell and lineage system. Am J Physiol. 1992 Aug;263(2 Pt 1):G139-48. doi: 10.1152/ajpgi.1992.263.2.G139.
[4]Lehallier B, Gate D, Schaum N, et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat Med. 2019 Dec;25(12):1843-1850. doi: 10.1038/s41591-019-0673-2.
[5] Jones DP, Mody VC Jr, Carlson JL, et al. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic Biol Med. 2002 Nov 1;33(9):1290-300. doi: 10.1016/s0891-5849(02)01040-7.
[6] Dall'Agnese A, Zheng MM, Moreno S, et al. Proteolethargy is a pathogenic mechanism in chronic disease. Cell. 2024 Nov 25:S0092-8674(24)01274-1. doi: 10.1016/j.cell.2024.10.051.
[7] Dance A. Live fast, die young. Nature. 2016 Jul 21;535(7612):453-5. doi: 10.1038/535453a.
[8] Genade T, Benedetti M, Terzibasi E, et al. Annual fishes of the genus Nothobranchius as a model system for aging research. Aging Cell. 2005 Oct;4(5):223-33. doi: 10.1111/j.1474-9726.2005.00165.x.
[9] Frenk S, Houseley J. Gene expression hallmarks of cellular ageing. Biogerontology. 2018 Dec;19(6):547-566. doi: 10.1007/s10522-018-9750-z.
[10] Matsuguma K, Hara T, Miyamoto D, et al. Improvement in aged liver regeneration using cell transplantation with chemically induced liver progenitors. J Hepatobiliary Pancreat Sci. 2024 Apr 3. doi: 10.1002/jhbp.1425.
[11] Yoshida Y. On some characteristics of the idioblast in Elodea leaf. J Fac Sci, Niigata Univ Ser. II 1958; 2: 173–178.
[12] Demanelis K, Jasmine F, Chen LS, et al. Determinants of telomere length across human tissues. Science. 2020 Sep 11;369(6509):eaaz6876. doi: 10.1126/science.aaz6876.
[13] MacKenzie KL, Franco S, May C, et al. Mass cultured human fibroblasts overexpressing hTERT encounter a growth crisis following an extended period of proliferation. Exp Cell Res. 2000 Sep 15;259(2):336-50. doi: 10.1006/excr.2000.4982.
[14] Martin JA, Mitchell CJ, Klingelhutz AJ, et al. Effects of telomerase and viral oncogene expression on the in vitro growth of human chondrocytes. J Gerontol A Biol Sci Med Sci. 2002 Feb;57(2):B48-53. doi: 10.1093/gerona/57.2.b48.
[15] Haussmann MF, Winkler DW, O'Reilly KM, et al. Telomeres shorten more slowly in long-lived birds and mammals than in short-lived ones. Proc Biol Sci. 2003 Jul 7;270(1522):1387-92. doi: 10.1098/rspb.2003.2385.
[16] Bilu Huang.Telomere DNA and ribosomal DNA co-regulation model for cell senescence[J].Negative,2021,12(3):9-15.doi:10.13276/j.issn.1674-8913.2021.03.003(2021).
[17] Yang F, Yi M, Liu Y, et al. Glutaredoxin-1 Silencing Induces Cell Senescence via p53/p21/p16 Signaling Axis. J Proteome Res. 2018 Mar 2;17(3):1091-1100. doi: 10.1021/acs.jproteome.7b00761.
[18] Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991 Sep;196(1):33-9. doi: 10.1016/0014-4827(91)90453-2.
[19] Henderson A.S., Warburton D., Atwood K.C. Location of ribosomal DNA in the human chromosome complement. Proc. Natl. Acad. Sci. USA. 1972;69:3394–3398. doi: 10.1073/pnas.69.11.3394.
[20] Fujita K, Horikawa I, Mondal AM, et al. Positive feedback between p53 and TRF2 during telomere-damage signalling and cellular senescence. Nat Cell Biol. 2010 Dec;12(12):1205-12. doi: 10.1038/ncb2123.
[21] Karni-Schmidt O, Friedler A, Zupnick A, et al. Energy-dependent nucleolar localization of p53 in vitro requires two discrete regions within the p53 carboxyl terminus. Oncogene. 2007 May 31;26(26):3878-91. doi: 10.1038/sj.onc.1210162.
[22] Lu KL, Nelson JO, Watase GJ, et al. Transgenerational dynamics of rDNA copy number in Drosophila male germline stem cells. Elife. 2018 Feb 13;7:e32421. doi: 10.7554/eLife.32421.
[23] Hoh J, Jin S, Parrado T, et al. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci U S A. 2002 Jun 25;99(13):8467-72. doi: 10.1073/pnas.132268899.
[24] Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010 Apr;45(4):286-90. doi: 10.1016/j.exger.2009.12.010.
[25] Stadler G, Rahimov F, King OD,et al. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat Struct Mol Biol. 2013 Jun;20(6):671-8. doi: 10.1038/nsmb.2571.
[26] Carlund O, Norberg A, Osterman P, et al. DNA methylation variations and epigenetic aging in telomere biology disorders. Sci Rep. 2023 May 16;13(1):7955. doi: 10.1038/s41598-023-34922-1.
[27] Larson K, Yan SJ, Tsurumi A, et al. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet. 2012 Jan;8(1):e1002473. doi: 10.1371/journal.pgen.1002473.
[28] Paredes S, Maggert KA. Ribosomal DNA contributes to global chromatin regulation. Proc Natl Acad Sci U S A. 2009 Oct 20;106(42):17829-34. doi: 10.1073/pnas.0906811106.
[29] Peterson EJ, Bögler O, Taylor SM. p53-mediated repression of DNA methyltransferase 1 expression by specific DNA binding. Cancer Res. 2003 Oct 15;63(20):6579-82.
[30] Challen GA, Sun D, Jeong M,et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011 Dec 4;44(1):23-31. doi: 10.1038/ng.1009.
[31] Liu L, Bailey SM, Okuka M, et al. Telomere lengthening early in development. Nat Cell Biol. 2007 Dec;9(12):1436-41. doi: 10.1038/ncb1664.
[32] Ren R, Deng L, Xue Y, et al. Visualization of aging-associated chromatin alterations with an engineered TALE system. Cell Res. 2017 Apr;27(4):483-504. doi: 10.1038/cr.2017.18.
[33]Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. Erratum in: Genome Biol. 2015 May 13;16:96. doi: 10.1186/s13059-015-0649-6.
[34] Foley NM, Hughes GM, Huang Z, et al. Growing old, yet staying young: The role of telomeres in bats' exceptional longevity. Sci Adv. 2018 Feb 7;4(2):eaao0926. doi: 10.1126/sciadv.aao0926.
[35] Stimpson KM, Sullivan LL, Kuo ME, et al. Nucleolar organization, ribosomal DNA array stability, and acrocentric chromosome integrity are linked to telomere function. PLoS One. 2014 Mar 24;9(3):e92432. doi: 10.1371/journal.pone.0092432.
[36] Iwama H, Ohyashiki K, Ohyashiki JH, et al. Telomeric length and telomerase activity vary with age in peripheral blood cells obtained from normal individuals. Hum Genet. 1998 Apr;102(4):397-402. doi: 10.1007/s004390050711.
[37]Robertson JD, Gale RE, Wynn RF, et al. Dynamics of telomere shortening in neutrophils and T lymphocytes during ageing and the relationship to skewed X chromosome inactivation patterns. Br J Haematol. 2000 May;109(2):272-9. doi: 10.1046/j.1365-2141.2000.01970.x.
[38] Lapham K, Kvale MN, Lin J, et al. Automated Assay of Telomere Length Measurement and Informatics for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics. 2015 Aug;200(4):1061-72. doi: 10.1534/genetics.115.178624.
[39]Stindl R. The Paradoxical Lengthening of Telomeres in Somatic Tissues of the Very Old: Aging Effect Meets Birth-Cohort Effect. J Exp Zool B Mol Dev Evol. 2016 Jun;326(4):213-4. doi: 10.1002/jez.b.22677.
[40] Leonida SRL, Bennett NC, Leitch AR, et al. Patterns of telomere length with age in African mole-rats: New insights from quantitative fluorescence in situ hybridisation (qFISH). PeerJ. 2020 Dec 4;8:e10498. doi: 10.7717/peerj.10498.
[41] Xu B, Li H, Perry JM, et al. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet. 2017 Jun 22;13(6):e1006771. doi: 10.1371/journal.pgen.1006771.
[42] Caratero A, Courtade M, Bonnet L, et al. Effect of a continuous gamma irradiation at a very low dose on the life span of mice. Gerontology. 1998;44(5):272-6. doi: 10.1159/000022024.
[43] Lou G, Palikaras K, Lautrup S, et al. Mitophagy and Neuroprotection. Trends Mol Med. 2020 Jan;26(1):8-20. doi: 10.1016/j.molmed.2019.07.002.
[44] Nijnik A, Woodbine L, Marchetti C, et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007 Jun 7;447(7145):686-90. doi: 10.1038/nature05875.
[45] Garza LA, Yang CC, Zhao T, et al. Bald scalp in men with androgenetic alopecia retains hair follicle stem cells but lacks CD200-rich and CD34-positive hair follicle progenitor cells. J Clin Invest. 2011 Feb;121(2):613-22. doi: 10.1172/JCI44478.
[46] Morrison, S., Wandycz, A., Akashi, K. et al. The aging of hematopoietic stem cells. Nat Med 2, 1011–1016 (1996). https://doi.org/10.1038/nm0996-1011
[47] Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010 Mar 25;464(7288):520-8. doi: 10.1038/nature08982.
[48] Rouhani FJ, Zou X, Danecek P,et al. Substantial somatic genomic variation and selection for BCOR mutations in human induced pluripotent stem cells. Nat Genet. 2022 Sep;54(9):1406-1416. doi: 10.1038/s41588-022-01147-3.
[49] Zhao T, Zhang ZN, Rong Z, et al. Immunogenicity of induced pluripotent stem cells. Nature 474, 212–215 (2011). doi.org/10.1038/nature10135.
[50] Ambrosi TH, Taheri S, Chen K, et al. Human skeletal development and regeneration are shaped by functional diversity of stem cells across skeletal sites. Cell Stem Cell. 2025 Mar 19:S1934-5909(25)00081-5. doi: 10.1016/j.stem.2025.02.013.
[51] Gladyshev VN, Anderson B, Barlit H, et al. Disagreement on foundational principles of biological aging. PNAS Nexus. 2024 Dec 3;3(12):pgae499. doi: 10.1093/pnasnexus/pgae499.
[52] Kobayashi T. Ribosomal RNA gene repeats, their stability and cellular senescence. Proc Jpn Acad Ser B Phys Biol Sci. 2014;90(4):119-29. doi: 10.2183/pjab.90.119.
[53] Mikuła-Pietrasik J, Pakuła M, Markowska M, et al. Nontraditional systems in aging research: an update. Cell Mol Life Sci. 2021 Feb;78(4):1275-1304. doi: 10.1007/s00018-020-03658-w.
[54] Sikder S, Baek S, McNeil T, et al. Centromere inactivation during aging can be rescued in human cells. bioRxiv [Preprint]. 2024 Sep 12:2023.12.30.573721. doi: 10.1101/2023.12.30.573721.
[55] Straight AF, Shou W, Dowd GJ, et al. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell. 1999 Apr 16;97(2):245-56. doi: 10.1016/s0092-8674(00)80734-5.
[56] Strahl-Bolsinger S, Hecht A, Luo K, et al. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 1997 Jan 1;11(1):83-93. doi: 10.1101/gad.11.1.83.
[57] Kobayashi T. Strategies to maintain the stability of the ribosomal RNA gene repeats--collaboration of recombination, cohesion, and condensation. Genes Genet Syst. 2006 Jun;81(3):155-61. doi: 10.1266/ggs.81.155.
[58] Grob A, Roussel P, Wright JE, et al. Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. J Cell Sci. 2009 Feb 15;122(Pt 4):489-98. doi: 10.1242/jcs.042382.
[59] Zhou X, Singh M, Sanz Santos G, et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021 Dec 1;11(12):3090-3105. doi: 10.1158/2159-8290.CD-20-1741.
[60] Liu X, Liu Z, Wu Z, et al. Resurrection of endogenous retroviruses during aging reinforces senescence. Cell. 2023 Jan 19;186(2):287-304.e26. doi: 10.1016/j.cell.2022.12.017.
[61] Partridge L, Fuentealba M, Kennedy BK. The quest to slow ageing through drug discovery. Nat Rev Drug Discov. 2020 Aug;19(8):513-532. doi: 10.1038/s41573-020-0067-7.
[62] Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007 Jan;8(1):35-46. doi: 10.1038/nrg2008.
[63] Yinfeng Xu1, Wei Wan.Spermidine inhibits rDNA transcription.Chinese Science Bulletin , Volume 69, Issue 15: 2072 - 2080 (2024) .https://doi.org/10.1360/TB-2024-0037.
[64] Li HY, Wang M, Jiang X, et al. CRISPR screening uncovers nucleolar RPL22 as a heterochromatin destabilizer and senescence driver. Nucleic Acids Res. 2024 Sep 11:gkae740. doi: 10.1093/nar/gkae740.
[65] Zarzycka W, Kobak KA, King CJ, et al.Hyperactive mTORC1/4EBP1 Signaling Dysregulates Proteostasis and Accelerates Cardiac Aging. bioRxiv [Preprint]. 2024 May 14:2024.05.13.594044. doi: 10.1101/2024.05.13.594044. Update in: Geroscience. 2024 Oct 9. doi: 10.1007/s11357-024-01368-w.
[66] Aldrich JC, Maggert KA. Transgenerational inheritance of diet-induced genome rearrangements in Drosophila. PLoS Genet. 2015 Apr 17;11(4):e1005148. doi: 10.1371/journal.pgen.1005148.
[67] Widjaja AA, Lim WW, Viswanathan S, et al. Inhibition of IL-11 signalling extends mammalian healthspan and lifespan. Nature. 2024 Aug;632(8023):157-165. doi: 10.1038/s41586-024-07701-9.
[68] Robinson PS, Coorens THH, Palles C, et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat Genet. 2021 Oct;53(10):1434-1442. doi: 10.1038/s41588-021-00930-y.
[69] de Bakker DEM, Valenzano DR. Turquoise killifish: A natural model of age-dependent brain degeneration. Ageing Res Rev. 2023 Sep;90:102019. doi: 10.1016/j.arr.2023.102019.
[70] Kabacik S, Lowe D, Fransen L, et al. The relationship between epigenetic age and the hallmarks of aging in human cells. Nat Aging. 2022 Jun;2(6):484-493. doi: 10.1038/s43587-022-00220-0.
[71] Ramunas J, Yakubov E, Brady JJ, et al. Transient delivery of modified mRNA encoding TERT rapidly extends telomeres in human cells. FASEB J. 2015 May;29(5):1930-9. doi: 10.1096/fj.14-259531.
[72] Muñoz-Lorente MA, Cano-Martin AC, Blasco MA. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat Commun. 2019 Oct 17;10(1):4723. doi: 10.1038/s41467-019-12664-x.
[73] Jaijyan DK, Selariu A, Cruz-Cosme R,et al. New intranasal and injectable gene therapy for healthy life extension. Proc Natl Acad Sci U S A. 2022 May 17;119(20):e2121499119. doi: 10.1073/pnas.2121499119. Epub 2022 May 10. Erratum in: Proc Natl Acad Sci U S A. 2022 Aug 30;119(35):e2212836119. doi: 10.1073/pnas.2212836119. Erratum in: Proc Natl Acad Sci U S A. 2023 Aug 8;120(32):e2311483120. doi: 10.1073/pnas.2311483120.
[74] Chew GL, Campbell AE, De Neef E, et al. DUX4 Suppresses MHC Class I to Promote Cancer Immune Evasion and Resistance to Checkpoint Blockade. Dev Cell. 2019 Sep 9;50(5):658-671.e7. doi: 10.1016/j.devcel.2019.06.011.
[75] Schratz KE, Flasch DA, Atik CC, et al. T cell immune deficiency rather than chromosome instability predisposes patients with short telomere syndromes to squamous cancers. Cancer Cell. 2023 Apr 10;41(4):807-817.e6. doi: 10.1016/j.ccell.2023.03.005.
[76] Atzmon G, Cho M, Cawthon RM, et al. Evolution in health and medicine Sackler colloquium: Genetic variation in human telomerase is associated with telomere length in Ashkenazi centenarians. Proc Natl Acad Sci U S A. 2010 Jan 26;107 Suppl 1(Suppl 1):1710-7. doi: 10.1073/pnas.0906191106.
[77] Willeit P, Willeit J, Mayr A, et al. Telomere length and risk of incident cancer and cancer mortality. JAMA. 2010 Jul 7;304(1):69-75. doi: 10.1001/jama.2010.897.
[78] Ocampo A, Reddy P, Martinez-Redondo P, et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell. 2016 Dec 15;167(7):1719-1733.e12. doi: 10.1016/j.cell.2016.11.052.
[79] Sahu SK, Reddy P, Lu J, et al. Targeted partial reprogramming of age-associated cell states improves markers of health in mouse models of aging. Sci Transl Med. 2024 Sep 11;16(764):eadg1777. doi: 10.1126/scitranslmed.adg1777.
[80] Grunewald M, Kumar S, Sharife H, et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science. 2021 Jul 30;373(6554):eabc8479. doi: 10.1126/science.abc8479.
[81] Gill D, Parry A, Santos F, et al. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. Elife. 2022 Apr 8;11:e71624. doi: 10.7554/eLife.71624.
[82] Fahy GM,Brooke RT,James P.,et al.Reversal of epigenetic aging and immunosenescent trends in humans[J].Aging Cell,2019,18(6):e13028.doi: 10.1111/acel.13028.
[83] Hanley BP, Brewer K, Church G. Results of a 5-Year N-of-1 Growth Hormone Releasing Hormone Gene Therapy Experiment. Rejuvenation Res. 2021 Dec;24(6):424-433. doi: 10.1089/rej.2021.0036.
[84] Njemini R, Abeele MV, Demanet C, et al. Age-related decrease in the inducibility of heat-shock protein 70 in human peripheral blood mononuclear cells. J Clin Immunol. 2002 Jul;22(4):195-205. doi: 10.1023/a:1016036724386.
[85] Corrigendum to: 17-a-estradiol late in life extends lifespan in aging UM-HET3 male mice; nicotinamide riboside and three other drugs do not affect lifespan in either sex. Aging Cell. 2022 Nov;21(11):e13672. doi: 10.1111/acel.13672. Epub 2022 Jul 27. Erratum for: Aging Cell. 2021 May;20(5):e13328. doi: 10.1111/acel.13328.
[86] Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004 Dec 17;306(5704):2105-8. doi: 10.1126/science.1101731.
[87] Brunquell J, Raynes R, Bowers P, et al. CCAR-1 is a negative regulator of the heat-shock response in Caenorhabditis elegans. Aging Cell. 2018 Oct;17(5):e12813. doi: 10.1111/acel.12813.
[88] Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008 Jun;10(6):676-87. doi: 10.1038/ncb1730.
[89] Zhou B, Kreuzer J, Kumsta C, et al. Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension. Cell. 2019 Apr 4;177(2):299-314.e16. doi: 10.1016/j.cell.2019.02.013.
[90] Li F, Zhu J, Liu J, et al. Targeting Estrogen Receptor Beta Ameliorates Diminished Ovarian Reserve via Suppression of the FOXO3a/Autophagy Pathway. Aging Dis. 2024 Feb 25. doi: 10.14336/AD.2024.0221.
[91] Li W, Zou Z, Cai Y, et al. Low-dose chloroquine treatment extends the lifespan of aged rats. Protein Cell. 2022 Jun;13(6):454-461. doi: 10.1007/s13238-021-00903-1. Erratum in: Protein Cell. 2024 Apr 1;15(4):313. doi: 10.1093/procel/pwad053.
[92] Wilhelm T, Byrne J, Medina R,et al . Neuronal inhibition of the autophagy nucleation complex extends life span in post-reproductive C. elegans. Genes Dev. 2017 Aug 1;31(15):1561-1572. doi: 10.1101/gad.301648.117.
[93] Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015 Jan 15;517(7534):302-10. doi: 10.1038/nature14190.
[94] Rodgers JT, Lerin C, Haas W, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005 Mar 3;434(7029):113-8. doi: 10.1038/nature03354.
[95] Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011 Feb 17;470(7334):359-65. doi: 10.1038/nature09787.
[96] Bonuccelli G, Brooks DR, Shepherd S, et al. Antibiotics that target mitochondria extend lifespan in C. elegans. Aging (Albany NY). 2023 Nov 9;15(21):11764-11781.doi:10.18632/aging.205229.
[97] Wang M, Zhang J, Qiu J, et al. Doxycycline decelerates aging in progeria mice. Aging Cell. 2024 Jul;23(7):e14188. doi:10.1111/acel.14188.
[98] Gao X, Yu X, Zhang C, et al. Telomeres and Mitochondrial Metabolism: Implications for Cellular Senescence and Age-related Diseases. Stem Cell Rev Rep. 2022
Oct;18(7):2315-2327.doi:10.1007/s12015-022-10370-8.
[99] Gibbons JG, Branco AT, Yu S, et al. Ribosomal DNA copy number is coupled with gene expression variation and mitochondrial abundance in humans. Nat Commun. 2014 Sep 11;5:4850.doi:10.1038/ncomms5850.
[100] Van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003 Dec 16;16(1):29-37. doi: 10.1152/physiolgenomics.00122.2003.
[101] Vermulst M, Bielas JH, Kujoth GC, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007 Apr;39(4):540-3. doi: 10.1038/ng1988.
[102] Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011 Nov 2;479(7372):232-6. doi: 10.1038/nature10600.
[103] Fang Y, Medina D, Stockwell R, et al. Sexual dimorphic metabolic and cognitive responses of C57BL/6 mice to Fisetin or Dasatinib and quercetin cocktail oral treatment. Geroscience. 2023 Oct;45(5):2835-2850. doi: 10.1007/s11357-023-00843-0.
[104] Lee E, Carreras-Gallo N, Lopez L, et al. Exploring the effects of Dasatinib, Quercetin, and Fisetin on DNA methylation clocks: a longitudinal study on senolytic interventions. Aging (Albany NY). 2024 Feb 22;16(4):3088-3106. doi: 10.18632/aging.205581.
[105] Xia X, Yang Y, Liu P, et al. The senolytic drug ABT-263 accelerates ovarian aging in older female mice. Sci Rep. 2024 Oct 5;14(1):23178. doi: 10.1038/s41598-024-73828-4.
[106] Facchini A, Mariani E, Mariani AR, et al. Increased number of circulating Leu 11+ (CD 16) large granular lymphocytes and decreased NK activity during human ageing. Clin Exp Immunol. 1987 May;68(2):340-7.
[107]Schaum N, Lehallier B, Hahn O, et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. 2020 Jul;583(7817):596-602. doi: 10.1038/s41586-020-2499-y.
[108] Newman LE, Weiser Novak S, Rojas GR, et al. Mitochondrial DNA replication stress triggers a pro-inflammatory endosomal pathway of nucleoid disposal. Nat Cell Biol. 2024 Feb;26(2):194-206. doi: 10.1038/s41556-023-01343-1.
[109] Sturmlechner I, Zhang C, Sine CC, et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science. 2021 Oct 29;374(6567):eabb3420. doi: 10.1126/science.abb3420.
[110] Raundhal M, Ghosh S, Myers SA,et al. Blockade of IL-22 signaling reverses erythroid dysfunction in stress-induced anemias. Nat Immunol. 2021 Apr;22(4):520-529. doi: 10.1038/s41590-021-00895-4. Epub 2021 Mar 22. Erratum in: Nat Immunol. 2021 Nov;22(11):1465. doi: 10.1038/s41590-021-01031-y.
[111] Cenini G, Sultana R, Memo M, et al. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic Biol Med. 2008 Jul 1;45(1):81-5. doi: 10.1016/j.freeradbiomed.2008.03.015.
[112] Cenini G, Sultana R, Memo M, et al. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer's disease. J Cell Mol Med. 2008 Jun;12(3):987-94. doi: 10.1111/j.1582-4934.2008.00163.x.
[113] Lanni C, Uberti D, Racchi M, et al. Unfolded p53: a potential biomarker for Alzheimer's disease. J Alzheimers Dis. 2007 Aug;12(1):93-9. doi: 10.3233/jad-2007-12109.
[114]Norheim KL, Ben Ezra M, Heckenbach I, et al. Effect of nicotinamide riboside on airway inflammation in COPD: a randomized, placebo-controlled trial. Nat Aging. 2024 Dec;4(12):1772-1781. doi: 10.1038/s43587-024-00758-1.
[115] Harrison DE, Strong R, Reifsnyder P, et al. 17-a-estradiol late in life extends lifespan in aging UM-HET3 male mice; nicotinamide riboside and three other drugs do not affect lifespan in either sex. Aging Cell. 2021 May;20(5):e13328. doi: 10.1111/acel.13328. Epub 2021 Mar 31. Erratum in: Aging Cell. 2022 Nov;21(11):e13672. doi: 10.1111/acel.13672.
[116] Kaiqiao Yang, Kazuya Nishiwaki, Hideaki Mizobata et al, The Greenland shark genome: insights into deep-sea ecology and lifespan extremes. bioRxiv 2025.02.19.638963; doi: https://doi.org/10.1101/2025.02.19.638963
[117][Li Y, Ma Y, Song L, et al . SIRT3 deficiency exacerbates p53/Parkin‑mediated mitophagy inhibition and promotes mitochondrial dysfunction: Implication for aged hearts. Int J Mol Med. 2018 Jun;41(6):3517-3526. doi: 10.3892/ijmm.2018.3555.
[118][Hoshino A, Mita Y, Okawa Y, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. doi: 10.1038/ncomms3308.
[119]Oka T, Hikoso S, Yamaguchi O, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012 May 10;485(7397):251-5. doi: 10.1038/nature10992.
[120] Smith P, Willemsen D, Popkes M, et al. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. Elife. 2017 Aug 22;6:e27014. doi: 10.7554/eLife.27014.
[121] Kundu P, Lee HU, Garcia-Perez I, et al. Neurogenesis and prolongevity signaling in young germ-free mice transplanted with the gut microbiota of old mice. Sci Transl Med. 2019 Nov 13;11(518):eaau4760. doi: 10.1126/scitranslmed.aau4760.
[122] Wilmanski T, Gibbons SM, Price ND. Healthy aging and the human gut microbiome: why we cannot just turn back the clock. Nat Aging. 2022 Oct;2(10):869-871. doi: 10.1038/s43587-022-00294-w.
[123] Ghosh TS, Shanahan F, O'Toole PW. Toward an improved definition of a healthy microbiome for healthy aging. Nat Aging. 2022 Nov;2(11):1054-1069. doi: 10.1038/s43587-022-00306-9.
[124] Leite G, Pimentel M, Barlow GM, et al. Age and the aging process significantly alter the small bowel microbiome. Cell Rep. 2021 Sep 28;36(13):109765. doi: 10.1016/j.celrep.2021.109765.
[125] Collins SL, Stine JG, Bisanz JE, et al. Bile acids and the gut microbiota: metabolic interactions and impacts on disease. Nat Rev Microbiol. 2023 Apr;21(4):236-247. doi: 10.1038/s41579-022-00805-x.
[126] Shalon D, Culver RN, Grembi JA, et al. Profiling the human intestinal environment under physiological conditions. Nature. 2023 May;617(7961):581-591. doi: 10.1038/s41586-023-05989-7.
[127] Kim DH, Lee JW. Tumor suppressor p53 regulates bile acid homeostasis via small heterodimer partner. Proc Natl Acad Sci U S A. 2011 Jul 26;108(30):12266-70. doi: 10.1073/pnas.1019678108.
[128] Chen P, Li D, Chen Y, et al. p53-mediated regulation of bile acid disposition attenuates cholic acid-induced cholestasis in mice. Br J Pharmacol. 2017 Dec;174(23):4345-4361. doi: 10.1111/bph.14035.
[129] Frommherz L, Bub A, Hummel E, et al. Age-Related Changes of Plasma Bile Acid Concentrations in Healthy Adults--Results from the Cross-Sectional KarMeN Study. PLoS One. 2016 Apr 19;11(4):e0153959. doi: 10.1371/journal.pone.0153959.
[130]Bertolotti M, Gabbi C, Anzivino C, et al. Age-related changes in bile acid synthesis and hepatic nuclear receptor expression. Eur J Clin Invest. 2007 Jun;37(6):501-8. doi: 10.1111/j.1365-2362.2007.01808.x.
[131] Wang W, Li N, Xu H, et al. ILC3s regulate the gut microbiota via host intestinal galactosylation to limit pathogen infection in mice. Nat Microbiol. 2025 Mar;10(3):654-666. doi: 10.1038/s41564-025-01933-9.
[132]Shuai Ma∙”Zhejun Ji,∙ Bin Zhang,et al.Spatial Transcriptomic Landscape Unveils Immunoglobin-associated Senescence as a Hallmark of Aging.cell.2024.
https://doi.org/10.1016/j.cell.2024.10.019
[133] Moon H, Jeong D, Choi JW, et al. Microplastic exposure linked to accelerated aging and impaired adipogenesis in fat cells. Sci Rep. 2024 Oct 13;14(1):23920. doi: 10.1038/s41598-024-74892-6.
[134] Kroemer G, Maier AB, Cuervo AM, et al. From geroscience to precision geromedicine: Understanding and managing aging. Cell. 2025 Apr 17;188(8):2043-2062. doi: 10.1016/j.cell.2025.03.011.
[135] Huang, Bilu, Demonstration of the Feasibility of Mainstream Aging Theories and Intervention (February 01, 2025). Available at SSRN: https://ssrn.com/abstract=5151210 or http://dx.doi.org/10.2139/ssrn.5151210
转载本文请联系原作者获取授权,同时请注明本文来自黄必录科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3440171-1491971.html?mobile=1
收藏