
 精选
精选
研究背景
硅负极具有3579 mAh g⁻1的高理论比容量,但在锂化/去锂化过程中巨大的体积变化(约300%)会导致材料结构破坏,表现出低功率密度和较差的循环稳定性。同时,硅电极表面上的SEI脆弱且不稳定,无法承受巨大体积变化,在循环过程中会不断破坏和重构。纳米化的硅虽然提高了离子传输性能,但较大比表面积和加剧的副反应会消耗电解液,导致容量快速衰减。高温下这些不良反应会加速,尤其在大电流密度下容易发生析锂,可能导致热失控,带来严重的安全风险。通过电解液调控和电极设计可实现特定组分/结构的稳定SEI,但如何在电极与电解液间构筑高效界面保护层仍是一个难题。近期,电极和电解液之间的相互作用引起了越来越多的关注。电解液添加剂可以提高电极的界面稳定性,其氧化还原路径具有多样性,同时电极材料表面的差异性导致反应活性与路径不同。过渡金属的特殊d轨道可催化电解液分解,通过调控官能团或引入杂原子可有效提升电化学活性。通过电极结构与电解液的协同设计,可使目标溶剂分子特异性吸附于电极表面的内亥姆霍兹面,进而优先分解形成特定组分的SEI。因此,亟需通过控制电极与电解质界面之间的相互作用,从而构建稳定的SEI。
Catalysis‑Induced Highly‑Stable Interface on Porous Silicon for High‑Rate Lithium‑Ion Batteries
Zhuobin Han, Phornphimon Maitarad, Nuttapon Yodsin, Baogang Zhao, Haoyu Ma, Kexin Liu, Yongfeng Hu, Siriporn Jungsuttiwong, Yumei Wang, Li Lu, Liyi Shi, Shuai Yuan, Yongyao Xia* and Yingying Lv*
Nano-Micro Letters (2025)17: 200
https://doi.org/10.1007/s40820-025-01701-8
本文亮点
1. 通过原位蚀刻与共生长的协同过程在多孔硅表面构建超薄富缺陷氧化物层。
2. 基于纳米级催化界面设计,催化电解液添加剂FEC可分解为富含LiF的SEI。
3. 多孔硅表面形成的致密SEI在不同温度下均表现出优异的高倍率循环稳定性。
内容简介
硅因其高能量密度而成为锂离子电池负极的关键材料。然而,传统的碳复合材料或纳米结构方法无法解决循环过程中固态电解质界面(SEI)大量形成带来的倍率性能差和有限的循环寿命等问题。复旦大学夏永姚、上海大学吕盈盈等人通过协同刻蚀和水解过程,在多孔硅表面原位构筑了一层纳米级超薄且均匀的氧化铝-氧化钛(ATO)催化界面。该富含缺陷的氧化物界面促进了氟乙烯碳酸酯(FEC)的选择性吸附,并催化其转化为LiF。由此产生的富含无机物的SEI层具有电化学稳定性,有利于离子传输,尤其是在高倍率循环和高温条件下。这种具有大比表面积的多孔硅(114 m2 g⁻1)被坚固的SEI层所保护,其初始库仑效率高达84.7%,即使在25 A g⁻1的高倍率下仍能保持692 mAh g⁻1的可逆容量,并且在5 A g⁻1下1000次循环中平均库仑效率高达99.7%,同时在50 ℃,2 A g⁻1下循环500次后的容量保持率达80.0%,显著优于同类型的硅负极材料。通过这种催化层构建的稳定SEI,为快充电池及高温下硅基负极的发展提供了一种借鉴思路。
图文导读
I p-Si@ATO负极材料的制备
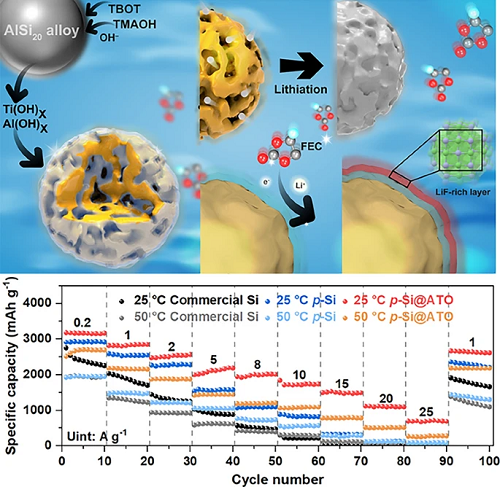
将微米级商业化AlSi₂₀合金超声分散于乙醇中,依次加入四甲基氢氧化铵(提供碱性环境)和15 mg钛酸四丁酯。AlSi₂₀合金中的金属铝和钛酸四丁酯同时在碱性环境下进行刻蚀和水解,生成无定形的氢氧化物,其进一步脱水交联,即可得到含有缺陷氧化铝-氧化钛(ATO)的多孔硅材料。
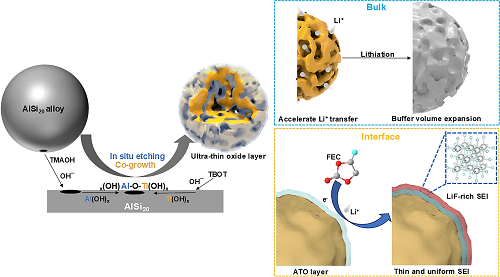
图1. p-Si@ATO的原位刻蚀与共生长合成示意图。
II p-Si@ATO负极材料的形貌与结构分析
形貌特征:p-Si@ATO保留AlSi₂₀的球形形貌,表面呈现数十纳米级开放孔道(图2a);TEM显示其具有中空枝状多孔结构(图2b及S4);
孔结构分析:氮气吸脱附等温线呈IV型特征(图2e),比表面积112.4 m2 g⁻1,孔径呈双模分布(介孔4.9 nm,大孔50-80 nm),源于铝的选择性蚀刻,可有效缓冲硅体积膨胀、提升传质速率和对电解液的吸附;
表面组成:HRTEM显示p-Si@ATO表面包覆3-5 nm非晶氧化物层(含Ti、Al、Si元素,图2c,d),其晶格条纹间距0.31 nm对应硅(111)晶面(图2d);组成成分:刻蚀后XRD谱中金属铝峰消失(图2f),ICP-OES定量表明p-Si@ATO的元素组成为85 wt% Si、5 wt% Al及0.3 wt% Ti。
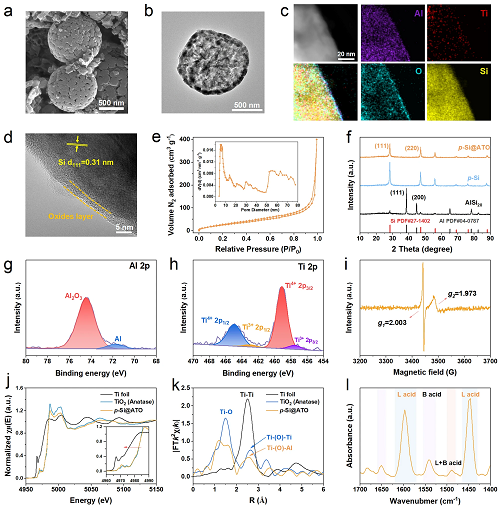
图2. p-Si@ATO的形貌与结构表征。(a) p-Si@ATO颗粒的SEM图像;(b) p-Si@ATO颗粒的TEM图像;(c, d) 表面氧化层的元素分布图和HRTEM图像;(e) p-Si@ATO的氮气吸附/脱附等温线及其相应的孔径分布;(f) p-Si@ATO、p-Si和AlSi₂₀合金的XRD图谱;(g, h) p-Si@ATO的高分辨率Al 2p和Ti 2p 的XPS;(i) 25 °C下p-Si@ATO粉末的EPR谱;(j) Ti箔、TiO₂和p-Si@ATO的归一化Ti K边的XAS;(k) k2加权EXAFS R空间的傅里叶变换;(l) 25 °C下p-Si@ATO粉末的Pyridine IR。
III p-Si@ATO负极材料的表面分析
表面化学成分:XPS分析证实p-Si@ATO表面存在氧化物层。Al 2p谱(图2g)在74.4 eV处显示Al₂O₃特征峰,71.7 eV处弱峰表明残留金属Al可能参与锂存储并提升电子传导性。Ti 2p谱(图2h)主峰位于459.0 eV(2p3/2)和464.7 eV(2p1/2),归属于Ti⁴⁺;次峰(457.2 eV和463.1 eV)表明存在Ti3⁺。EPR谱(图2i)证明了在g=2.003和1.973处存在氧空位和Ti3⁺信号,二者协同增强电子传导与负电荷吸附能力。
Ti3⁺形成机制:对比实验表明,以纯硅(不含Al)替代AlSi₂₀合金时,EPR仅检测到氧空位(g=2.002)而无Ti3⁺信号。碱性条件下金属Al的蚀刻与TBOT水解生成氢氧化物,通过Al-O-Ti键合形成非晶氧化物层。同步辐射XAS分析(图2j)显示Ti K边能量略低于锐钛矿TiO₂,证实Ti价态降低(Ti3⁺存在)。EXAFS谱(图2k)中1.5 Å处Ti-O键峰及2.5 Å处峰位偏移(因Al原子半径小于Ti)也证实Al-O-Ti键的存在。
表面酸性位点分析:吡啶吸附红外光谱(图2l)显示:1596.8 cm⁻1和1446.4 cm⁻1处吸收峰对应Lewis酸位点(L),1542.8 cm⁻1处弱峰为Brønsted酸位点(B),1488.8 cm⁻1处为L/B混合酸位点。Al-O-Ti键的形成增加配位不饱和金属位点,显著增强Lewis酸性。这些酸性位点和表面缺陷促进电解液吸附与催化分解,协同Ti3⁺及氧空位提升电子传导,结合介孔结构(加速传质并缓冲体积膨胀),共同构筑稳定SEI界面。
IV p-Si@ATO负极材料的电化学性能
不同温度下的首效:p-Si@ATO在25°C/50°C下0.2 A g⁻1首圈库伦效率(ICE)分别为84.7%/73.2%,显著高于商业硅(79.2%/70.2%)及纯多孔硅(71.6%/61.3%)(图S14)。三者的比表面积分别为112.4 m2 g⁻1, 98.5 m2 g⁻1 和10.1 m2 g⁻1。由于有效的界面保护,尽管p-Si@ATO的比表面积较大,但是其ICE值也较高。
循环稳定性:
(1)常温性能:5 A g⁻1下循环1000次,p-Si@ATO容量保持率79.8%(p-Si 41.1%,商业化硅13.7%,),平均库伦效率99.7%(图3a);2 A g⁻1下循环500次容量保持率99.5%(图S15)。
(2)高温性能:50 °C下5 A g⁻1循环500次后容量保持率67.5%(图3b),2 A g⁻1下循环500次容量保持率80.0%(图S16)。
高倍率性能:25 A g⁻1下p-Si@ATO可逆容量达692 mAh g⁻1(25 °C)与270 mAh g⁻1(50 °C),远超商业化硅(15 A g⁻1下容量趋近于零)(图3c)。并且p-Si@ATO可以在10、15和20 A g⁻1的高电流密度下实现稳定的长循环(图3d)。
实际应用验证:高负载量的p-Si@ATO电极可达3.2 mAh cm−2的面容量(图S17)。p-Si@ATO||LiFePO₄软包电池未预锂化条件下50次循环容量保持率77.4%,平均库伦效率97.5%(图3e、S18)。不同温度下和混合石墨的全电池如图S19和S20所示。
动力学特性:
(1)锂离子扩散系数:GITT测试表明p-Si@ATO的Li⁺扩散系数(~10⁻11 cm2 s⁻1)较商业硅(10⁻13 cm2 s⁻1)提升2个数量级,归因于介孔结构加速传质及表面活性位点促进吸附(图3f)。
(2)界面阻抗分析:DRT分析(图3g-i)揭示:p-Si@ATO的SEI阻抗(RSEI)与电荷转移阻抗(Rct)相较于p-Si和商业化硅显著降低,尤其在0.1-0.5 V去锂化阶段,τ₃(RSEI)与τ₄(Rct)峰强度减弱,证实其SEI具备快速去锂化动力学特性,有利于提升其可逆容量和倍率性能。
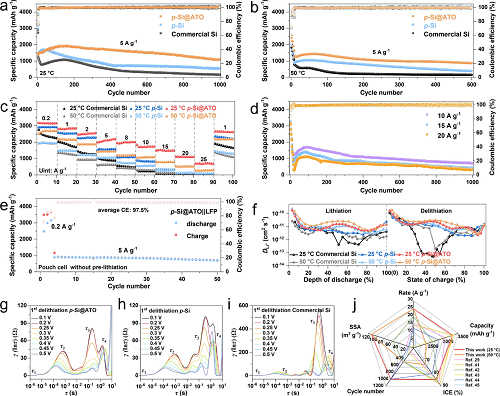
图3. p-Si@ATO、p-Si及商业硅负极的电化学性能对比。(a, b) p-Si@ATO、p-Si与商业硅在25 ℃和50 ℃下的长循环性能;(c) 三种电极在不同电流密度下的倍率性能;(d) p-Si@ATO在25 ℃下的高倍率循环性能;(e) p-Si@ATO||LiFePO₄软包全电池(未预锂化)在25 ℃下的循环性能;(f) 循环10次后三种电极的GITT;(g-i) 25 ℃下三种电极在初始充电过程中的DRT;(j) p-Si@ATO与文献报道的硅基负极材料性能对比。
V p-Si@ATO负极材料的电极与界面演化分析
循环后极片的形貌与厚度:循环100后颗粒表面的HRTEM显示p-Si@ATO的SEI相较于p-Si和商业化硅更薄且更均匀(图4a,b和图S23),这有利于硅颗粒均匀地发生锂化。p-Si@ATO循环10圈后的电极膨胀率(63.6%)则显著低于商业硅(140.0%),证实多孔结构有效抑制体积变化(图4c-f)。
富无机SEI形成过程:在首圈初始放电阶段(2.0-0.5 V),LSV中的多个还原峰对应电解液分解(图4g);准原位XPS追踪显示,2.0 V时F 1s谱便已经出现LiF峰(685 eV),随电压降低LiF的相对含量增加(图4h)。原位拉曼证实了在1.8 V之前便已经在416.6 cm⁻1出现了LiF的拉曼信号,并且硅的特征峰(518.0 cm⁻1)逐步减弱,表明SEI逐步在硅颗粒表面构建(图4i)。这表明在首圈放电过程中,硅颗粒的内部在更高的电压下优先生成了以LiF为主的SEI。由于LiF的化学稳定性,其在长循环过程中可以减少副反应,提升电极的循环稳定性。
SEI的动态演化过程:不同循环圈数后的XPS分析表明,p-Si@ATO电极中的LiF相对含量随循环从90.6 at%逐步增至93.1 at%(图2.33),表明SEI层通过动态重组趋于稳定,LiF持续生成并主导界面成分。C 1s谱中Li2CO₃(289.8 eV)含量先升后降,表明其作为初始SEI无机组分生成后部分分解,形成稳定无机-有机复合结构。而商业化硅LiF的相对含量含量从79.3 at%降至54.5 at%,同时含磷副产物(LixPOyFz、LixPFy)显著增加(687.0/688.5 eV),反映了电极表明的SEI在持续破裂-再生,导致电解液过度消耗与产气。并且Li₂CO₃含量持续上升,是因为体积变化导致活性表面持续暴露,引发电解液(如EC)反复分解,产气(CO₂)致SEI变得疏松多孔。
对比了三种电极循环100圈后的ToF-SIMS。商业硅SEI表层以有机物(C₂H₂O⁻信号主导)为主,LiF仅在内层富集。并且商业化硅电极的Si−信号在离子刻蚀100秒后方才显现,同时伴随C₂H₂O⁻信号的持续衰减。这表明其表面存在较厚的以有机成分为主导的SEI层,且随着深度增加,Li₂CO₃含量逐渐升高而LiF的相对强度有限。p-Si和p-Si@ATO电极在刻蚀初期即检测到明显的Si−信号,其中p-Si@ATO电极表现出更为突出的LiF₂⁻信号强度及明显较低强度的C₂H₂O⁻含量。由此可知,p-Si@ATO电极的外层SEI层厚度较商用硅电极更薄,其界面结构呈现"外疏内密"的梯度特征:外层为稀疏的有机薄层(C₂H₂O⁻信号强度更低),内层则为富含LiF的无机致密层(LiF₂⁻信号强度更高)。这种独特的结构源于ATO表面对FEC溶剂的选择性吸附及其催化分解机制。通过促进LiF的优先成核有效抑制了有机组分的无序堆积,避免发生更多的副反应。
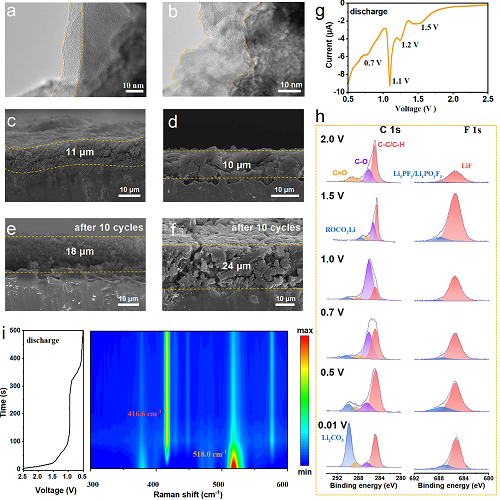
图4. p-Si@ATO负极的界面与电极分析。(a, b) 循环100次后p-Si@ATO (a) 和商业化硅(b) 电极的HRTEM图像;(c-f) p-Si@ATO(c, d)和商业硅(e, f) 电极的横截面SEM图像;(g) p-Si@ATO在0.05 mV s⁻1下的初始放电过程LSV;(h) 不同电压下p-Si@ATO电极的准原位XPS;(i) p-Si@ATO电极在0.2 A g⁻1下的首次放电过程原位电化学拉曼光谱。
VI 理论计算
溶剂吸附与分解优先级:FEC因LUMO能级最低(−0.37 eV,图5c),在初始锂化阶段优先还原分解,主导SEI内层组分。ATO表面对FEC的吸附能(−2.09 eV)显著低于SiO₂(−1.38 eV)与Al₂O₃(−1.72 eV),表明其更强的FEC富集能力(图5d)。
LiF形成路径优化:FEC在ATO表面吸附后的的C-F键长(1.41 Å)较SiO₂(1.45 Å)更易断裂,形成Li-F键后键长扩展至3.88-4.95 Å(图5e)。LiF在ATO界面形成的终态能量(−7.99 eV)较Al₂O₃(−7.75 eV)与SiO₂(−3.01 eV)更低,证实其在热力学更容易在电极表面生成LiF(图5f)。
电子结构调控:分波态密度(PDOS)分析表明,ATO的d带中心(1.52 eV)更高,较SiO₂(p带中心−0.58 eV)与Al₂O₃(p带中心−2.72 eV)表现出更高催化活性(图5g)。FEC吸附导致ATO的p带与d带上移(图S28),增强活性中间体吸附,促进LiF生成。
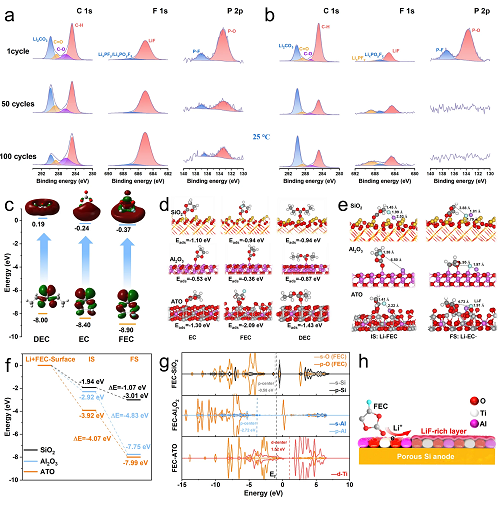
图5. (a, b) p-Si@ATO (a) 和商业硅 (a) 在25 °C下的C 1s、F 1s和P 2p XPS谱图。所有XPS均在100%放电深度(SOC)下使用拆解的半电池电极测量;(c) 溶剂分子的电子跃迁及Egap;(d) 溶剂分子在不同表面上的吸附构型;(e, f) LiF形成结构及其在SiO₂、Al₂O₃和ATO表面上的相对能量分布;(g) FEC在SiO₂、Al₂O₃和ATO表面上的PDOS;(h) FEC溶剂在ATO表面上形成LiF的界面催化示意图。
VII 结论
本研究针对硅负极的界面和体相问题,通过原位生长和刻蚀工艺在AlSi₂₀合金微球上制备了具有催化作用的纳米级超薄氧化物层的多孔硅微球。通过控制活性物质的缺陷态表面与电解液的相互作用,显著提升了FEC向LiF转化的反应活性。同时介孔结构增强了FEC的积聚效应,缓冲了硅负极的体积膨胀,加快了离子传输速率,保持了电极结构的完整性。电化学测试表明,p-Si@ATO电极在0.2 A g⁻1下的ICE为84.7%,在5 A g⁻1下循环1000次后容量保持率为79.8%。即使在25 A g⁻1的高电流密度下,其可逆容量仍可达692 mAh g⁻1,并在10、15和20 A g⁻1的高电流密度下实现了1000次稳定循环。结构分析表明,多孔结构有效缓解了硅负极的体积膨胀,保持了电极的宏观完整性。表征和理论计算揭示了p-Si@ATO表面衍生的SEI层富含LiF,且更薄、更均匀。LiF的形成电压高于有机SEI和Li2CO₃,其化学惰性、电子绝缘性和高机械强度显著减少了界面副反应,维持了电极的循环稳定性。这些特性共同赋予了p-Si@ATO电极优异的电化学性能。
作者简介

吕盈盈
本文通讯作者
上海大学 副教授
▍主要研究领域
多孔纳米材料/柔性器件的可控制备,及锂钠离子电池方向应用:(1)纳米尺寸多孔复合材料设计;(2)柔性多孔储能薄膜或器件制备。
▍主要研究成果
上海大学理学院纳米科学与技术研究中心副教授,博士生导师。上海市青年东方学者,上海市海外领军。研究方向为多孔储能材料及器件。先后在Advanced Materials,ACS Nano,Nano Letters等期刊发表SCI论文30余篇,被引2500余次,H因子18, i10指数为21。
▍Email:yyinglv@shu.edu.cn

夏永姚
本文通讯作者
复旦大学 教授
▍主要研究领域
电化学、化学电源。自1990年起一直从事电化学储能材料和储能技术的基础和应用研究,包括锂 (钠) 离子电池(包括有机、水系和固态)、电化学电容器和新型储能体系等。
▍主要研究成果
1987年毕业于浙江师范大学化学系获理学学士学位,1990年于吉林大学化学系获理学硕士学位,1997年于日本国佐贺大学应用化学系能源材料科学专业获工学博士学位。曾任中国化学会电化学专业委员会第七届主任,国际电池领域顶尖杂志Journal of Power Sources 编辑(editor)。主持包括国家重点研发计划(首席科学家),科技部“973计划”,“863计划”项目、国家自然科学基金杰出青年、重点、面上项目、上海市科委和企业合作项目等30余项。先后入选教育部新世纪优秀人才计划,上海市优秀学科带头人。
▍Email:yyxia@fudan.edu.cn
撰稿:原文作者
编辑:《纳微快报(英文)》编辑部
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2023 JCR IF=31.6,学科排名Q1区前3%,中国科学院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
转载本文请联系原作者获取授权,同时请注明本文来自纳微快报科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3411509-1486671.html?mobile=1
收藏