
 精选
精选
研究背景
太阳能与天然热液系统结合将革新CO₂加氢化学,但缺乏高效低成本催化剂体系仍是重大挑战。本文创新性地选择可太阳能循环再生的Zn为还原剂,Co为催化剂,在热液环境中实现了CO₂完全甲烷化。蜂窝状ZnO纳米片在Co表面原位生长形成Co@ZnO新型催化剂,通过ZnO辅助的CoOₓ还原机制抑制催化剂失活。稳定化Co-ZnO界面协同作用实现CO₂完全转化为CH₄。原位热液红外光谱证实甲酸为关键中间体,有效规避CO生成路径。本研究为高效CO₂转化与催化剂合成提供了一体化解决方案,推动了太阳能驱动甲烷化技术的发展。
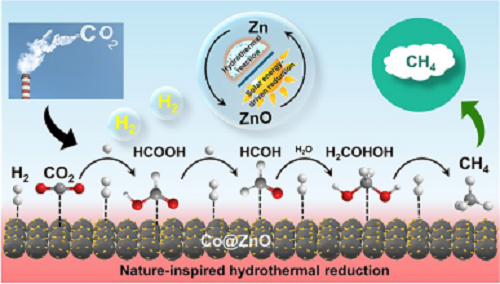
100% Conversion of CO2–CH₄ with Non‑Precious Co@ZnO Catalyst in Hot Water
Yang Yang, Xu Liu, Daoping He*, Fangming Jin*
Nano-Micro Letters (2025)17: 216
https://doi.org/10.1007/s40820-025-01711-6
本文亮点
1. 性能飞跃:开发了原位生成的Co@ZnO催化剂,通过Zn氧化提供的强还原环境与ZnO辅助CoOₓ还原机制,实现CO₂至CH₄的100%选择性转化。
2. 技术革新:Co-ZnO界面协同作用增强了CO₂吸附活化能力,并通过调控反应路径完全抑制CO生成,并成功抑制钴基催化剂失活。
3. 应用前景广阔:该工艺将太阳能驱动金属还原与水热催化相结合,无需外部供氢或复杂催化剂设计,为碳中和能源系统提供了高效、可持续的解决方案。
内容简介
太阳能与天然热液系统的结合将革新CO₂加氢化学,但缺乏高效且低成本的催化体系仍是挑战。上海交通大学金放鸣等人选择可太阳能循环再生的Zn作为还原剂,以Co为催化剂,实现了高效水热CO₂甲烷化。蜂窝状ZnO纳米片在Co表面原位生长,形成Co@ZnO催化剂,通过ZnO辅助的CoOₓ还原抑制钴失活。稳定的Co-ZnO界面协同作用实现了CO₂至CH₄的完全转化。原位水热红外光谱证实甲酸为中间体,从而避免了CO生成及副反应路径。本研究为高效CO₂转化与催化剂合成提供了一体化方案,为太阳能驱动的CO₂甲烷化开辟了新途径。
图文导读
I Zn与Co协同水热CO₂甲烷化
在250–325 °C范围内考察了Zn-Co体系水热CO₂甲烷化性能。仅使用Zn的对照实验中仅有甲酸生成,而加入金属Co后显著提成了CH₄的产率。反应后Zn氧化为ZnO。为排除ZnO催化效应,以ZnO为催化剂时仍仅生成甲酸。单独Co催化时CH₄产率仅约40%,归因于CO₂溶解受限。逐步提高溶液碱度可使CH₄产率提升至77%(图1a),但过量碱度导致产率下降,呈现火山型关系。通过分析不同碱度下碳物种(H₂CO₃、HCO₃⁻、CO₃2⁻)分布,发现CH₄产率与HCO₃⁻浓度正相关,13CO₂同位素实验证实了CH₄碳源来自CO₂还原(图1b)。
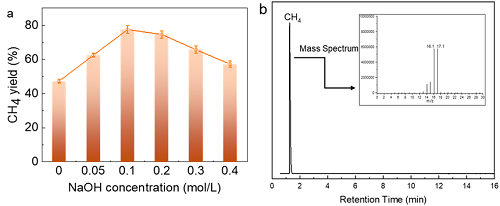
图1. (a) NaOH浓度对CH₄收率的影响, (b) 13CO2同位素反应后气态产物的GC-MS分析。
固体产物XRD分析(图2a)显示:41.7°、44.8°、47.6°、62.7°、75.9°归属于六方密堆积钴(PDF#05–0727),31.7°、34.4°、36.3°、47.5°、56.6°、62.9°、68.0°对应于ZnO(PDF#36–1451),表明Zn氧化为ZnO产氢,而Co保持零价态。SEM显示催化剂表面生长有纳米片包覆的纳米棒结构(图2b,c),高倍图像显示蜂窝状纳米片(厚度~10 nm)。EDX面扫(图2d–g)表明纳米棒为Co,包覆层为ZnO,证实Co@ZnO结构。超声去除ZnO后(图S2a),XRD(图S2b)确认剩余Co保持金属态。HRTEM(图2h)显示Co棒具有规则晶格条纹(d=2.04 Å,对应Co(111)晶面),边缘ZnO的d=2.8 Å对应纤锌矿ZnO(100)晶面。Co-ZnO界面处IFFT图像显示无序晶格,表明Co@ZnO异质结构成功构建。通过时间分辨实验追踪ZnO在Co表面的生长过程,初始阶段Co棒表面光,反应10–30 min后ZnO纳米片开始形成,1–3 h后继续生长。CH₄产量在30 min至1 h内急剧上升并持续累积,表明Co@ZnO催化剂的形貌演变显著提升甲烷生成。
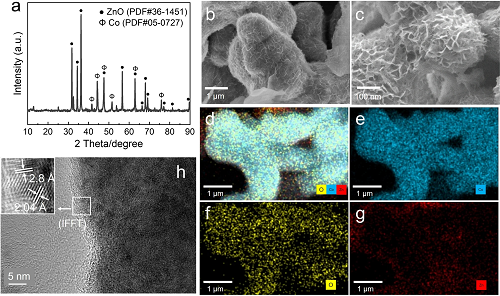
图2. Co@ZnO催化剂表征:a) XRD谱;b,c) SEM图像;d-g) SEM-EDS分析;h) HRTEM图像及IFFT分析。
II Zn-Co水热体系中Co零价态维持
XPS分析(图3a)显示Co@ZnO与预还原商业Co(Co–H₂)表面均存在CoO与Co共存,CoO信号可能源于空气氧化。通过原位水热红外监测金属氧化过程发现:Fe+Co体系在250 °C下5 min出现Co–O信号,Al+Co体系检测到弱信号,而Zn+Co体系无Co–O信号,表明Zn的强还原氛围抑制Co氧化。H₂-TPR分析(图3b)显示商业Co在235 °C(Co₃O₄→CoO)和300 °C(CoO→Co)出现还原峰,而Co@ZnO仅出现单峰,表明ZnO促进CoOₓ还原。物理混合Co与ZnO的TPR谱与商业Co相似,说明Co-ZnO直接接触是氢溢流增强的关键。以H₂替代Zn为还原剂时,商业Co催化CH₄产率较Co@ZnO低52%且生成CO、乙酸等副产物,而Co@ZnO仅生成CH₄及微量甲酸。钴基催化剂表面CoOₓ易催化CO₂→CO转化并引发羰基插入生成乙酸,而Co@ZnO维持零价态有效抑制了副反应。ICP分析显示反应液钴溶出量仅8.57 ppm,表明催化剂稳定性优异。
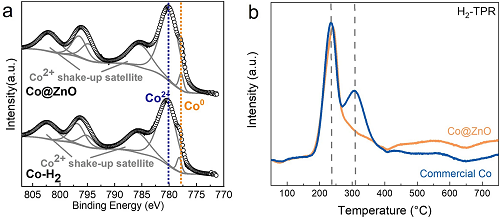
图3. Co@ZnO与商业Co的(a)XPS及(b)H₂-TPR对比。
通过XAFS(图4a–d)分析Co@ZnO界面结构:Co K边XANES显示吸收边介于CoO与Co之间,表明界面Co呈部分氧化态(0<δ<2)。EXAFS拟合表明Co–O–Zn/Co配位结构形成。CO₂-TPD(图4e)显示Co@ZnO中强/中/弱吸附位点脱附峰强度均高于商业Co,且中强吸附位点(370–600 °C)丰度更高,有利于甲烷化反应。DFT计算(图4f)表明Co@ZnO表面CO₂吸附能(–0.27 eV)显著低于纯Co(0.32 eV),归因于ZnO诱导的电子重分布增强了CO₂吸附稳定性。
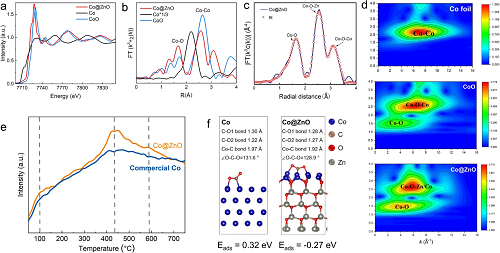
图4. 同步辐射XAFS分析:(a) Co K边XANES;(b) EXAFS傅里叶变换谱;(c) EXAFS拟合曲线;(d) 小波变换分析;(e) CO₂-TPD谱;(f) DFT计算对比。
III 水热CO₂甲烷化反应机理
实验结果中产物检测表明气相产物以CH₄为主,无CO检出;液相仅含痕量甲酸。原位红外(图5a)显示:反应2 min开始生成甲酸,25 min出现甲醛特征峰(2920、2840 cm⁻1)45 min后3014 cm⁻1处CH₄特征峰强度显著增加甲酸作为中间体呈现生成-消耗动态平衡,表明其逐步转化为甲醛并最终生成CH₄。以甲酸或CO单独作为反应物时,甲酸生成CH₄的产率显著高于CO,验证了甲酸路径的高效性。仅使用Zn时,甲酸虽快速生成但无CH₄产出;ZnO催化体系亦仅累积甲酸,表明Co对深度还原至关重要。结合DFT计算(图5b),Co@ZnO表面HCOOH生成路径的决速步能垒(0.53 eV)低于CO路径(0.68 eV),说明甲酸路径更易进行。此外,Co-ZnO界面强金属-载体相互作用(SMSI)通过电子调控进一步优化了CO₂吸附与活化。基于上述结果,提出反应机理(图5c):水热条件下Zn氧化产H₂并生成ZnO,H₂经ZnO活化后参与CO₂还原。CO₂首先被吸附活化,经两步加氢生成甲酸;甲酸进一步加氢脱水形成甲醛中间体,最终深度加氢生成CH₄。ZnO作为氢缓冲层促进H₂解离,Co表面高效氢溢流加速中间体转化,同时界面电子效应抑制CO路径,实现CH₄的100%选择性。
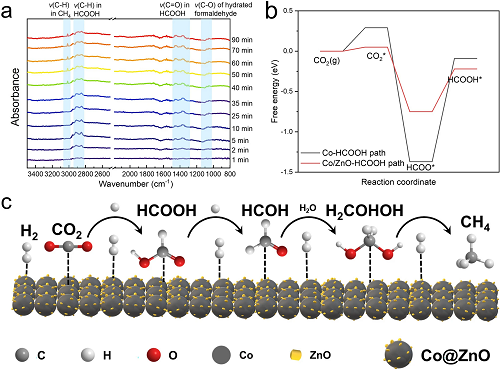
图5. (a)原位水热红外光谱;(b) 甲酸路径反应能垒;(c) 反应机理示意图。
IV 工艺优化与能量平衡
通过调节Zn/Co摩尔比、温度和时间优化反应条件。当Zn用量增至90 mmol(300 °C,2 h)时,CH₄产率达100%(图6)。催化剂循环5次或延长反应至10 h后,Co@ZnO结构保持稳定。对比其他过渡金属(Fe、Ni、Cu)及贵金属催化剂(Pd/C、Pt/C),其CH₄产率均低于30%,凸显Co@ZnO体系的优越性。能量平衡分析表明,每摩尔反应可回收535.628 kJ能量。经三次反应循环后,加热能耗可完全补偿,后续反应可实现净能量输出。这一结果表明该工艺具备规模化应用潜力。
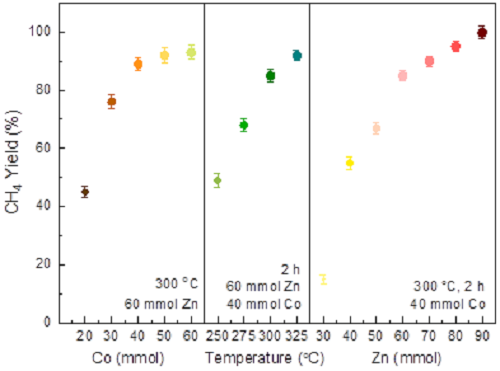
图6. 工艺参数优化对甲烷产率影响结果。
V 总结
本研究报道了基于Zn和Co的简单金属体系实现水热CO₂甲烷化。值得注意的是,在Zn提供的强还原氛围及ZnO活化原位H的作用下,Co始终维持金属态。Co表面原位形成蜂窝状ZnO纳米片,构建了Co@ZnO催化剂。零价Co与Co-ZnO相互作用通过完全抑制CO中间体生成、增强CO₂及甲酸中间体吸附,实现了CO₂还原CH₄的100%产率。这种整合太阳能与水热萨巴蒂埃反应的Zn-Co体系推动了钴基催化剂在化工领域的应用,为高效、可持续的CO₂甲烷化提供了新思路。
作者简介

金放鸣
本文通讯作者
上海交通大学 特聘教授
▍主要研究领域
水热(太阳能协同)转化二氧化碳为碳一或多碳产物;水热转化生物质废弃物、湿垃圾产高附加值产物;非生物成因有机合成同深海微生物生命过程相互关系;水热转化有机废弃物为高附加值产品的工业化等。
▍主要研究成果
金放鸣,上海交通大学(环境科学与工程学院)特聘教授,博士生导师,学校学术委员会委员。清华大学固体废物处理与环境安全教育部重点实验室学术委员会副主任;日本东北大学环境科学研究院Fellow(5名中唯一的亚洲人);日本东北大学客座教授;日本理化学研究所客座研究员;Japan Prize(素称日本诺奖,诺奖得主J. Goodenough和吉野彰曾获此奖)官方提名人;Scientific Reports和Energy Science & Engineering杂志编委,Low Carbon Energy, Environment and Management Journal副主编。长期以来主要从事生物质(包括有机废弃物)和CO2的水热资源化利用的研究,在PNAS, JACS, Angew, Energy & Environmental Science, Green Chemistry, Environmental Science & Technology, AIChE Journal, Chem comm等高质量国际知名学术杂志发表学术论文近300篇,60多项高创新专利。
▍Email:fmjin@sjtu.edu.cn
撰稿:原文作者
编辑:《纳微快报(英文)》编辑部
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2023 JCR IF=31.6,学科排名Q1区前3%,中国科学院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
转载本文请联系原作者获取授权,同时请注明本文来自纳微快报科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3411509-1485954.html?mobile=1
收藏