
 精选
精选
研究背景
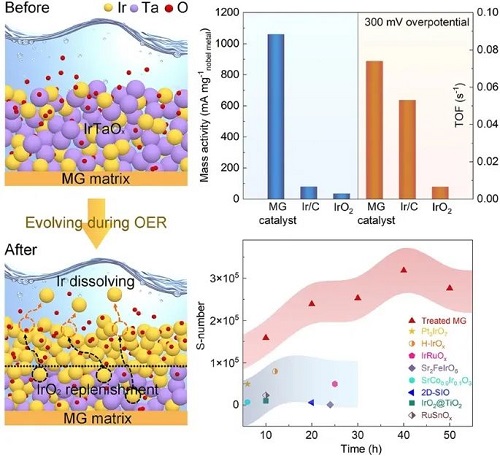
质子交换膜(PEM)电解水技术是绿氢制备的可靠途径,然而PEM电解水中的析氧反应(OER)催化剂仍面临贵金属负载量高、质量活性低、耐久性不足等局限,这阻碍了其进一步产业化。因此,为了提高 PEM 电解水的竞争力,仍需进一步开发具有更高活性、长期稳定和更低 Ir 含量的新型酸性 OER 电催化剂。近日西北工业大学李睿副教授和北京科技大学刘雄军、吕昭平教授等联合报道了一种以 Ir-Ta 基非晶合金为基体自构建的 IrO₂/IrTaOₓ双层纳米结构催化剂用于高效酸性OER,为PEM电解水制氢提供了新方案。
Durable Acidic Oxygen Evolution Via Self‑Construction of Iridium Oxide/Iridium‑Tantalum Oxide Bi‑Layer Nanostructure with Dynamic Replenishment of Active Sites
Qi Guo, Rui Li*, Yanan Zhang, Qiqin Zhang, Yi He, Zhibin Li, Weihong Liu, Xiongjun Liu*, Zhaoping Lu*
Nano-Micro Letters (2025)17: 165
https://doi.org/10.1007/s40820-025-01680-w
本文亮点
1. 以Ir-Ta基非晶合金为基体原位构建IrO₂/IrTaOx双层纳米结构,可用于高效酸性OER催化剂。
2. Ir和Ta间电子相互作用调控催化剂活性位点的配位性质,从而产生了优越的酸性OER活性和稳定性。
3. 催化剂中非晶态IrTaOx层通过向内晶化和选择性溶解动态补充表面Ir活性位点的消耗,确保长期OER耐久性。
内容简介
西工大李睿&北科刘雄军&吕昭平等人报告了一种用于酸性OER的自构建层状催化剂,该催化剂通过直接使用Ir-Ta基非晶合金作为基体,于OER过程中在非晶IrTaOx纳米结构上原位形成了纳米多孔IrO₂表面。这种独特的结构大大提高了Ir的可及性和利用率,在300 mV的过电位下实现了1.06 A mgIr ⁻¹的高活性,分别是商用Ir/C和IrO₂的13.6倍和31.2倍。该催化剂在酸性大电流密度下也表现出极佳的稳定性,这表明它具有实际应用的潜力。实验分析表明,通过Ir和Ta之间的电子相互作用,有效调节了表面活性Ir物种的配位性质,防止它们迅速演化为高价态,并抑制了晶格氧的参与。此外,底层非晶态IrTaOx通过向内晶化和选择性溶解动态地补充了表面活性位点的消耗,从而确保了催化剂的长期OER耐久性。
图文导读I 催化剂制备和性能表征
Ir30Ta35Ni29Nb6非晶合金(MG)基体是采用真空甩带法制备的 (记为As-spun MG)。随后对其进行表面酸处理,在MG基体上形成了催化活性表面 (记为Treated MG)。然后在0.5 M H₂SO₄电解液中评估了As-spun MG和Treated MG的OER性能,并与商用Ir/C和IrO₂进行比较。图1a中的LSV曲线表明,Treated MG在电流密度为10 mA cm⁻²时的过电位为211 mV(电流归一化为几何表面积),明显低于其前驱体合金(901 mV)和商用催化剂(Ir/C为312 mV,IrO₂为330 mV)。图1b中相应的塔菲尔斜率为37.5 mV dec⁻¹,这进一步证明了Treated MG具有优异的电化学反应动力学。此外,这些特征值还与最近报道的各种Ir基OER电催化剂进行了比较,与这些贵金属基催化剂相比,Treated MG表现出了卓越的酸性OER活性。如图1d所示,在过电位为300 mV时,Treated MG的周转频率 (TOF) 值高于商用Ir/C和IrO₂,这表明催化Ir位点具有更高的内在活性。特别的是,作为一种自支撑材料,该催化剂表面在300 mV的过电位下具有1.06 A mgIr⁻¹的显著质量活性,分别是商用Ir/C(78.7 mA mgIr⁻¹)和IrO₂(34.3 mA mgIr⁻¹)的13.6倍和31.2倍(图1d),这表明该催化剂不仅具有更高的OER活性,而且还通过减少Ir的用量而具有成本优势。
除了高电催化活性外,催化剂的运行稳定性也是PEM电解水的一个重要指标。在100 mA cm⁻²的恒定电流密度下,使用计时电位测试评估了该催化剂在酸中的OER耐久性。如图1e所示,在超过650小时的时间里,Treated MG的性能退化可以忽略不计,过电位退化率约为10 μV h⁻¹,低于PEM电解槽的最新规格(14 μV h⁻¹)。此外,论文还采用了S-number来评估催化剂的化学稳定性,该参数被定义为氧分子数与溶解的 Ir 阳离子数之比。如图1f所示,经计算,Treated MG在初始状态下(≤ 50 h)的S-number高达2.7×10⁵,优于最近报道的许多Ir基电催化剂。这一结果表明,Treated MG在OER过程中保持很高的稳定性。
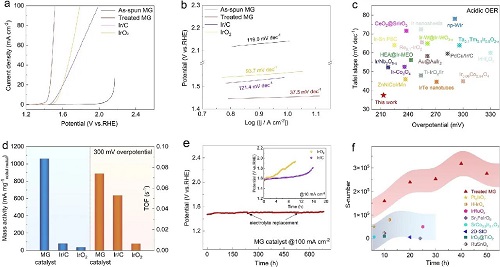
图1.(a) 在0.5 M H₂SO₄电解液中的线性极化曲线;(b) 相应的Tafel斜率;(c) 在10 mA cm⁻²时的过电位和Tafel斜率与最近报道的Ir基酸性OER电催化剂的比较;(d) 在300 mV过电位下的质量活性和周转频率;(f) 在50-h OER稳定性测试期间,Treated MG与最近报道的各种电催化剂的S-number的比较。
II 结构演变分析
图 2a 展示了Treated MG的横截面透射电子显微镜(TEM)图像。经酸处理后,在非晶合金基体上形成了厚度约为100 nm的蚀刻层。图2b中的HR-TEM以及相应的选区电子衍射(SAED)图显示,该蚀刻层具有非晶态性质,这也与GI-XRD的表征结果一致(图2c)。TEM-EDS曲线显示,催化剂表面的Ni和Nb元素被选择性地蚀刻掉,从而形成了IrTaOx层状纳米结构(图2d)。此外,在非晶态IrTaOx层表面还形成了厚度约为15 nm的Ta₂O₅钝化层。在酸处理过程中逐渐形成的Ta₂O₅钝化层可保护IrTaOx层和非晶基体免受过度腐蚀,从而稳定了层状催化剂结构。STEM元素图谱分析进一步证实了非晶基体上复合氧化物层的形成和元素分布(图2e)。

图2.(a) Treated MG横截面的TEM图像(顶层为聚焦离子束沉积的铂纳米颗粒);(b) 在 (a) 中蓝色方框标记的Treated MG层结构的HRTEM图像,插图显示了黄色方框标记区域的SAED图样;(c) 催化电极中IrTaOx非晶层的XRD图谱;(d) 非晶态IrTaOx层的TEM-EDS分析图谱;(e) Treated MG横截面的HAADF-TEM图像和相应的EDS元素图谱。
论文认为,非晶态IrTaOx纳米结构是Treated MG催化剂优异OER性能的原因。为了验证这一假设,论文研究了催化剂在长期酸性OER过程中的结构演变。如表面形貌随反应时间的变化所示(图 3a),IrTaOx表面逐渐形成了纳米多孔结构,并稳定为平均尺寸约为100 nm的双连续纳米多孔。即使在400小时后,纳米多孔结构仍几乎保持不变,这表明其在酸性OER催化过程中具有超强的稳定性。图3b和3c分别展示了IrTaOx非晶层在经过50小时和200小时的OER催化反应后的变化,并通过TEM对其进行了详细的表征分析。值得注意的是,在OER催化过程中,Treated MG展现出了一种独特的双层结构。随着OER的持续进行,IrTaOx非晶层上方逐渐形成了一层新的薄表面层。在OER进行50小时后,该薄表面层可以被观察到,厚度大约为11 nm左右。在OER进行200小时后,该层表现出明显的纳米多孔结构,这与上述SEM的观察结果一致。此外,在OER反应200小时后,该表面层的厚度也有所增加,并在随后的长期OER催化过程中保持稳定,其厚度大约为23 nm。此外,通过HRTEM图像还观察到有相当数量的新生纳米晶体分散在表面IrO₂多孔层下方的IrTaOx非晶层中(见图3c右下方插图所示),这表明非晶态IrTaOx层在OER过程中进行了一定程度的晶化。通过对产生纳米晶的IrTaOx层进行SAED测试分析,其产生的多晶环对应于IrO₂晶体的(312)、(211)、(210)和(101)晶面,证实了在IrTaOx非晶层中新生纳米晶体为IrO₂纳米晶。此外,IrTaOx层的厚度在长期OER后有所减少(即在OER进行200小时后减少了约24 nm),这进一步证实了表面IrO₂纳米晶体主要来自下面的IrTaOx层。对Treated MG在长期OER过程中的Ir元素溶解情况进行ICP测试,鉴于在OER过程中表面Ir活性物种不可避免地会发生溶解,论文推断,IrTaOx非晶层通过向层内晶化和选择性溶解起到了储层的作用,使新形成的IrO₂纳米晶体能够动态地补偿表面活性IrO₂的损失。这种机制提供了一种补充能力,有助于保持催化电极表面活性和稳定性的平衡。
图3d进一步说明了这一动态演化过程,主要包括组成元素的扩散、氧化和溶解。在强酸性和强氧化条件下,氧原子在Treated MG的IrTaOx非晶层大量积累,在化学势梯度的驱动下,氧原子进一步向内扩散。与此同时,非贵金属Ta元素因其耐腐蚀性能相对低于Ir元素而优先向外溶解,导致表面出现大量空位。因此,由于化学势梯度,Ir原子倾向于向外层扩散和聚集以填补这些空位,并通过与氧结合形成IrO₂晶体,从而演变为观察到的双层结构。随着OER的持续进行,无定形的IrTaOx层将会进一步内部晶化和动态结构演化,特别是对表面活性物质Ir的溶解做出响应。根据以上实验数据可以看出,在Ir30Ta35Ni29Nb6MG基体上自构建的纳米多孔双层结构对于提高OER催化性能至关重要,其中催化电极表面生成的IrO₂纳米晶体可作为有效的催化活性位点,而纳米级多孔结构则提供了高比表面积,有利于电荷的快速转移。此外,位于催化电极表面IrO₂多孔层下方的无定形IrTaOx层可以通过晶化和选择性溶解动态演化出IrO₂纳米晶体,补充表面Ir活性位点的消耗,平衡酸性介质中的OER活性和稳定性。这种结构的形成不仅增强了催化电极的活性和稳定性,而且为设计新型高效OER催化剂提供了新的思路。

图3. Treated MG在酸性OER期间的动态结构演变:(a) 酸性OER条件下表面形成纳米多孔IrO₂结构的原位SEM图像;(b) 酸性OER 50小时后IrTaOx层结构的TEM图像;(c) 酸性OER 200小时后IrTaOx层结构的TEM图像。插图显示了(c)中所选区域的相应HRTEM图像(黄色方框:右上方;蓝色方框:右下方);(d) OER期间IrTaOx层结构演变和表面IrO₂补充示意图。
III 电子结构与反应机理分析
使用高分辨率XPS对Treated MG催化电极的表面化学状态进行了检测。图4a显示了在0~200小时的酸性OER过程中,Treated MG催化电极表面的Ir 4f光谱变化情况。分析结果显示,Treated MG在OER进行50小时后,与初始状态相比,其表面的金属Ir已完全转化为氧化物。具体而言,Ir的氧化态主要归因于Irᴵⱽ和Irᴵᴵᴵ物种,它们的特征峰分别位于62.1/65.1 eV和63.0/65.8 eV。此外,特别值得注意的是,在Treated MG经过100小时的阳极极化后,表面Ir的结合能发生了正向移动,变化范围在0.2~0.3 eV之间,这种结合能的正向移动可能促进了Irᴵᴵᴵ向Irᴵⱽ的进一步转化。随着OER进行至200小时,Irᴵⱽ和Irᴵᴵᴵ物种的峰面积比出现了显著变化,即Irᴵᴵᴵ物种已完全转化为了更高价态的Irᴵⱽ物种。此外,Ir 4f的光谱数据揭示了Irᴵⱽ的结合能在200-h OER后有所下降,其Irᴵⱽ峰右移了0.2~0.3 eV左右。这一变化表明,反应形成的Irᴵⱽ物种是稳定的,不会进一步过度氧化成更高的氧化态(即Irⱽ和Irⱽᴵ氧化物)。事实上,最近的研究表明,酸性OER触发Ir溶解的途径主要涉及可溶性Irᴵᴵᴵ或高价Ir(>Irᴵⱽ)物种的形成。因此,该论文认为表面稳定的Irᴵⱽ物种是催化电极化学稳定性高的关键因素。
论文还深入研究了Ta 4f光谱的变化特征。如图4b所示,XPS峰值的差异清晰地揭示了在酸性OER过程中金属Ta⁰向氧化态Taⱽ的完全转变。此外,与初始状态相比,Taⱽ的结合能发生了约0.6~0.7 eV的正向移动,这一现象表明在OER过程中,Ta与Ir的电子调控行为,可能通过改变Ir的电子结构来优化其催化活性,进一步增强了催化电极的整体性能。Ir与Ta之间的强烈电子相互作用促使Ta向高价态氧化,这一过程能够有效吸附多余的氧原子,从而防止具有催化活性的Irᴵⱽ氧化物在酸性OER过程中进一步氧化为更高、更不稳定的价态,例如Irⱽ或Irⱽᴵ物种。这种作用显著提升了催化电极的耐久性,因为它阻断了可能导致催化电极失活的反应路径。通过Ir和Ta的电子协同效应为催化电极在严苛的酸性OER条件下的长期稳定性提供了保障。
图4c详细展示了在酸性OER过程中O 1s XPS光谱的演变情况。通过对催化电极表面的氧物种进行精确的化学状态分析,核心级光谱能够被解析为三种不同的氧物种:催化电极表面吸附的H₂O、催化电极表面吸附的OH⁻以及催化电极的晶格氧。进一步地,为了区分与不同金属结合的晶格氧,论文观察到位于531.0 eV处的峰被确定为与Ta结合的氧(Ta-O),而530.1 eV附近的峰则被认为是与Ir结合的氧(Ir-O)。在这种情况下,电负性较低的Ta原子预计会优先与氧中间体结合形成Ta-O键,这一行为能够最大限度地减少Ir的电子损失,并有效抑制催化电极表面的活性Irᴵⱽ氧化物进一步氧化为可溶的高价对应物,例如不稳定的Irⱽ或Irⱽᴵ物种。这种Ta-O键的优先结合行为对于维持催化电极的稳定性和活性至关重要,因为它减少了活性位点的损失,从而提高了催化电极的耐久性。此外,经过长时间的OER测试,观察到催化电极晶格氧与表面吸附OH⁻的峰面积比有所增加。这一变化说明在OER过程中,Treated MG催化电极表面氧物种的动态变化和相互作用,进一步证实了Ta和Ir元素在催化剂中与氧相互作用的电子环境变化。晶格氧与吸附OH⁻的峰面积比的增加可能与催化电极表面结构的重构和元素电子结构的调整有关,这一趋势揭示了催化电极表面吸附的氧中间体不易受到质子化的影响,而更倾向于与电解质中的H₂O发生反应,形成过氧化氢(OOH⁻)中间产物,随后产生O₂。这种反应路径表明,在OER催化过程中吸附质机制(AEM)在催化过程中占主导地位,相较于晶格氧机制(LOM)更为有利。
通过实验对Treated MG在OER过程中的反应途径进行验证,论文利用同位素标记技术,并配合操作DEMS测量(图3d)。该方法可以精确追踪反应过程中的氧同位素变化,从而直接观察和确认OER过程中的氧中间体行为。随着OER电流的增加,原位质谱检测到了气态氧产物,包括³²O₂(¹⁶O¹⁶O)、³⁴O₂(¹⁶O¹⁸O)和³⁶O₂(¹⁸O¹⁸O),如图4e所示。值得注意的是,在OER电位范围内,³⁴O₂与³²O₂的信号比维持在约0.53%的较低水平,这一现象表明在催化电极表面并没有显著的LOM发生。这意味着在OER过程中,吸附在催化电极表面的氧中间体更倾向于通过AEM参与反应,而不是通过晶格氧直接参与。在对比Treated Ir30Ta35Ni29Nb6 MG催化电极与未添加Ta元素的Treated Ir30Ni42Nb28 MG催化电极时,论文发现前者生成的³⁴O₂/³²O₂值显著低于后者(图4f)。这一显著差异表明,在IrO₂/IrTaOx催化表面上,晶格氧的参与被有效抑制。这种抑制作用可能源于Ir和Ta之间的强电子相互作用,这种相互作用不仅优化了催化电极的电子结构,还降低了LOM中晶格氧的氧化和迁移,从而提高了电化学稳定性。
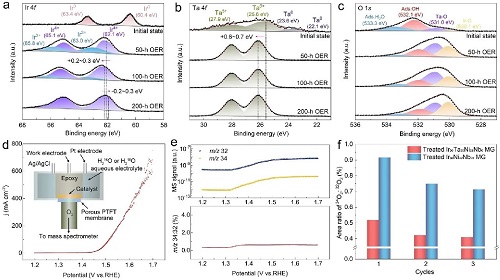
图4. 长期酸性OER稳定性测试期间的XPS分析和同位素标记的原位DEMS表征:(a) Ir 4f、(b) Ta 4f和 (c) O 1s的XPS光谱;(d) 用于DEMS测试的Treated MG催化电极的CV周期。插图显示了原位DEMS的示意图;(e) 顶部:在CV循环期间收集到的³²O₂ (¹⁶O + ¹⁶O) 和³⁴O₂ (¹⁶O + ¹⁸O) 的DEMS信号;底部:随着电位的增加,³⁴O₂/³²O₂的质谱峰面积比;(f) 在H₂¹⁶O/0.5 M H₂SO₄电解液里的3个OER周期中,Treated Ir30Ta35Ni29Nb6 MG和Treated Ir30Ni42Nb28 MG催化电极的³⁴O₂/³²O₂比值。
为了深入探究Ta与Ir之间的电子调制行为对催化电极稳定性的影响,论文进行了DFT计算。图5a清晰地展示了 (Ir, Ta)O₂和IrO₂ (110) 晶面的电荷密度分布情况,揭示了Ta原子在这一结构中扮演电子供体的角色。进一步的Bader电荷分析结果提供了更详细的电子结构变化信息:Ta的掺杂导致Ir原子的供电子能力从-1.601 e减少到-1.375 e,而O原子的受电子能力则从0.799 e增加到了1.121 e(图5b)。这一观察结果表明,加入Ta原子后,Ir阳离子的价态降低,可能会增强Ir在酸性环境中的抗溶解性。此外,Ir-O的共价性降低导致形成了额外的Ta-O键。所形成的Ir-O-Ta局域结构中的强电子相互作用有助于形成更稳定的表面构型,这可能会进一步抑制OER期间晶格氧的参与,从而提高催化电极的稳定性。图5c展示了 (Ir, Ta)O₂和IrO₂结构的投影态密度(PDOS)分析结果。通过引入Ta原子,费米能级附近的总态密度出现了增加,这一现象表明Ta原子有效地调节了Ir的d电子特性。这种调节作用优化了反应Ir位点的局部配位环境,对于催化电极的活性具有重要影响。此外,还观察到Ta原子和O原子的轨道能级逐渐升高,费米能级附近的杂化最强。这一结果进一步支持了Ta-O键的优先形成。论文还计算了IrO₂和 (Ir, Ta)O₂在零外加电势 U=0 V(标准电势)下的OER自由能图。对于 (Ir, Ta)O₂和IrO₂而言,速率决定步骤 (RDS) 被确定为从*O到*OOH的转化(图5d)。(Ir, Ta)O₂的能垒降低到了2.277 eV,明显低于IrO₂(2.618 eV)。这表明 (Ir, Ta)O₂中的Ir位点是有效的活性位点,能极大地促进水氧化反应动力学。这种促进作用主要归因于Ir和Ta之间的电子协同效应,它通过下移d带中心优化了O中间产物的吸附强度,从而调节了OER中间产物的吸附能。
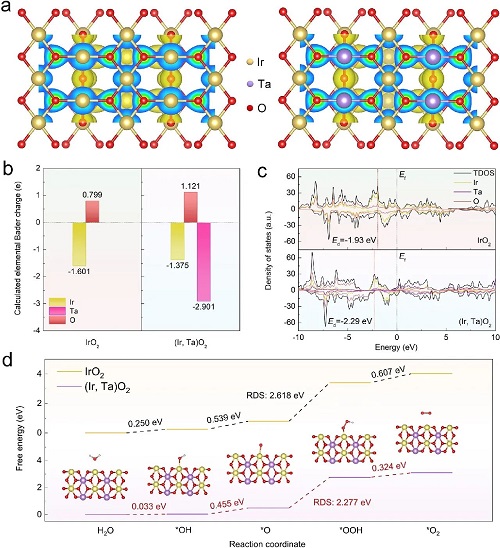
图5. DFT计算:(a) (Ir, Ta)O₂和IrO₂模型 (110) 晶面的电荷密度分布;(b) IrO₂和 (Ir, Ta)O₂结构的Bader电荷分布;(c) 计算出的IrO₂和 (Ir, Ta)O₂的PDOS。Ef代表费米能级,Ed代表d带中心;(d) (Ir, Ta)O₂和IrO₂模型的自由能图。
IV 总结
该论文基于Ir-Ta-Ni-Nb MG成功开发了一种具有Ir-Ta基非晶氧化物纳米层状结构的催化电极。在酸性OER过程中,IrTaOx非晶层表面可以原位生成具有高催化活性的IrO₂纳米多孔层,形成的IrO₂/IrTaOx双层结构显著增加了催化电极表面活性位点的数量,并促进了电子的快速转移。同时底部的IrTaOx纳米结构能够通过内部晶化和溶解动态演化成IrO₂纳米晶体,有效补充了表面活性位点的消耗,从而维持了催化电极的长期服役稳定性。因此,这种可以实现自构建的IrO₂/IrTaOx双层催化电极在300 mV的过电位的质量活性高达1.06 A mgIr⁻¹,并在100 mA cm⁻²的电流密度下提供了超过650小时的长期OER稳定性。电子结构分析和DEMS测试结果共同揭示了Ir与Ta之间的强电子相互作用能够有效平衡活性Ir物种的价态,防止形成高价可溶物种,同时也抑制了OER过程中晶格氧的参与,从而实现活性和稳定性的同步提升。这项工作为稳定活性Ir氧化物以实现长期酸性OER提供了新的视角,也为开发适用于实际PEM电解水应用的高效耐用催化剂开辟了一条新途径。
作者简介

李睿
本文通讯作者
西北工业大学 副教授
▍主要研究领域
多主元复杂合金的设计、研发及其功能特性研究,探索非晶合金、高熵合金、纳米晶合金等亚稳态合金以及三维纳米多孔金属在清洁能源储存及转换领域的应用。
▍个人简介
西北工业大学材料学院副教授、博士生导师。主要从事多主元复杂合金的设计、研发及其功能特性研究,探索非晶合金、高熵合金、纳米晶合金等亚稳态合金以及三维纳米多孔金属在清洁能源储存及转换领域的应用。相关研究成果在Advanced Materials,Nature Communications,Advanced Function Materials和Acta Materialia等期刊发表论文30余篇,授权国家发明专利8项。
▍Email:ruili@nwpu.edu.cn

刘雄军
本文通讯作者
北京科技大学 研究员
▍主要研究领域
非晶合金、高熵合金和无序合金能源材料等前沿金属材料结构与性能的多尺度计算模拟与合金设计。
▍个人简介
刘雄军,北京科技大学新金属材料国家重点实验室研究员,博士生导师。主要从事非晶合金、高熵合金和无序合金能源材料等前沿金属材料结构与性能的多尺度计算模拟与合金设计研究工作。在Nature、Nature Communications、Advanced Materials、Physical Review Letters和Acta Materialia等国际学术期刊上发表SCI论文190余篇,被引用12500余次;授权国家发明专利40余件;获国家自然科学二等奖2项、教育部自然科学一等奖和二等奖各1项。
▍Email:xjliu@ustb.edu.cn

吕昭平
本文通讯作者
北京科技大学 特聘教授
▍主要研究领域
新型高性能钢铁材料、块体非晶合金及高熵合金。
▍个人简介
吕昭平,北京科技大学副校长,教育部长江学者特聘教授,国家杰出青年科学基金获得者;国际学术杂志Intermetallics主编。长期从事新型高性能钢铁材料、块体非晶合金及高熵合金等方面的研究工作,承担国家自然基金委重大、重点项目和重大国际合作项目、973项目等多项国家研究课题;在Nature、Science、等学术刊物上发表论文230余篇,引用10500余次,授权中国发明专利30余项,美国发明专利2项,成功转化1项。1项成果入选2017年中国科学十大进展,以第一完成人获得国家自然科学二等奖2项,教育部自然科学一等奖和二等奖各1项。
▍Email:luzp@ustb.edu.cn
撰稿:原文作者
编辑:《纳微快报(英文)》编辑部
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2023 JCR IF=31.6,学科排名Q1区前3%,中国科学院期刊分区1区Top期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
转载本文请联系原作者获取授权,同时请注明本文来自纳微快报科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3411509-1481404.html?mobile=1
收藏