


背景介绍
一氧化碳(CO)的过度排放对空气质量构成了严重的环境威胁,引起了人们的广泛关注。铂(Pt)基催化剂因其独特的电子结构和优异的催化性能,被广泛应用于CO排放控制。然而,贵金属Pt的稀缺性和高成本阻碍了其广泛应用,特别是对于催化剂使用量需求大的工业烟气治理领域。因此,开发低负载、高性能的Pt基催化剂对于烟气CO治理至关重要。优化Pt物种配位结构是强化催化剂氧化活性的重要途径。本研究通过N2处理诱发Pt单原子迁移聚集形成全暴露Pt团簇,显著强化了催化剂在Pt负载量为0.1 wt.%下的催化活性。具体而言,通过NaOH蚀刻Ti-Si复合材料制备缺陷TiO2载体,随后使用浸渍法将Pt原子锚定在缺陷载体上,合成Pt单原子缺陷催化剂,然后在400 ℃下N2气氛中进行处理,以调节Pt物种的分散状态和配位环境,制备得到全暴露Pt团簇催化剂。活性评价结果表明,全暴露Pt团簇催化剂CO氧化活性得到显著提高,最低完全氧化温度相对未处理催化剂降低70 ℃,在130 ℃即可实现CO完全氧化。AC-TEM和XANFS等表征技术证实,全暴露Pt团簇催化剂上的Pt物种保持了最大的原子利用率,同时还含有多原子金属位点。DFT计算表明,与未处理催化剂上的单个Pt原子相比,N2处理的催化剂上全暴露的Pt团簇对CO和O2的吸附性能显著增强,这归因于Pt-Pt配位的存在提供了额外的吸附位点。
图文解读
1. 催化剂CO氧化性能评价
采用不同气氛(N2、O2和H2)在400 ℃下处理Pt单原子缺陷催化剂,结果如图1所示。结果显示,经过N2和H2处理的Pt/Ti-D H2和PtFEC/Ti-D N2表现出最好的CO氧化性能,而Pt/Ti-D O2表现出最差的催化活性。Pt/Ti-D H2和PtFEC/Ti-D N2均在130 ℃实现CO完全氧化,而Pt/Ti-D O2在220 ℃才能达到100%转化。结果表明,H2和N2处理对提高催化剂的CO氧化活性具有关键作用。进一步研究了4种样品的表观活化能(Ea)。与活性测试结果一致,Pt/Ti-D H2和PtFEC/Ti-D N2的表观活化能显著低于PtSA/Ti-D和Pt/Ti-D O2的表观活化能,表明H2和N2处理有利于优化活性中心和加速CO的氧化过程。尽管Pt/Ti-D H2和PtFEC/Ti-D N2表现出相同的催化活性,但考虑到N2的成本低廉和安全性,重点关注了PtFEC/Ti-D N2的催化性能和结构性质。
为了验证N2处理是否对常规Pt/Ti催化剂的活性具有促进作用,比较了在400 ℃下用N2处理PtSA/Ti-D催化剂与Pt/Ti催化剂的CO氧化性能。两种催化剂在N2处理后的氧化活性均显著增加。与Pt/Ti N2相比,PtFEC/Ti-D N2催化剂由于存在缺陷结构而表现出更好的催化活性。除气体种类外,考察了处理温度对催化剂氧化活性的影响。N2处理温度分别为300、400、500和600 ℃时,400 ℃处理的样品表现出最高的氧化活性,这归因于不同处理温度形成的Pt团簇大小不同。

图1.(a)PtSA/Ti-D、PtFEC/Ti-D N2、Pt/Ti-D O2和Pt/Ti-D H2的CO转化曲线;(b)不同催化剂上的表观活化能(Ea);(c)PtSA/Ti-D、PtFEC/Ti-D N2、Pt/Ti和Pt/Ti N2的CO转化曲线;(d)在N2气氛下不同温度处理1 h后PtSA/Ti-D的CO氧化活性。
2. 催化剂结构表征
采用球差电镜技术对PtSA/Ti-D和PtFEC/Ti-D N2催化剂表面Pt物种的形貌进行了详细分析。AC-HAADF STEM图像显示,Pt在PtSA/Ti-D催化剂表面高度分散,以单原子形式存在(图2a)。相比PtSA/Ti-D,N2处理后的PtFEC/Ti-D N2的TEM图像显示,催化剂表面存在明显的Pt团簇(图2b)。结果表明,N2处理可能通过诱导Pt原子的迁移和聚集,优化了催化剂的活性位点分布,从而提升了其催化性能。为了探究Pt团簇的原子层厚度,以K位点的Pt单原子作为参考,分析了从A点到B点的线强度分布。如图2c所示,Pt团簇中多数Pt原子的线强度与K位点的Pt单原子相当,这表明Pt团簇的厚度主要以单原子层为主,基本完全暴露在缺陷载体表面。此外,高分辨率TEM图像和EDS映射图进一步证实了Pt团簇在PtFEC/Ti-D N2催化剂表面的存在(图2d-g)。基于PtSA/Ti-D和PtFEC/Ti-D N2催化剂的AC-HAADF STEM图像结果,推导出PtFEC/Ti-D N2催化剂表面全暴露Pt团簇的形成过程。如图2h所示,在缺陷载体的制备阶段,通过调控TiO2的缺陷结构,形成富含缺陷的Ti-D载体。随后,通过浸渍法将Pt前驱体溶液引入载体中,Pt原子优先锚定在Ti-D的缺陷位置,形成单原子分散的PtSA/Ti-D催化剂。催化剂在400 ℃的N2气氛中处理中,Pt单原子的配位结构变得不稳定,Pt-O键发生断裂,导致Pt原子从缺陷位置脱离并迁移形成Pt团簇。
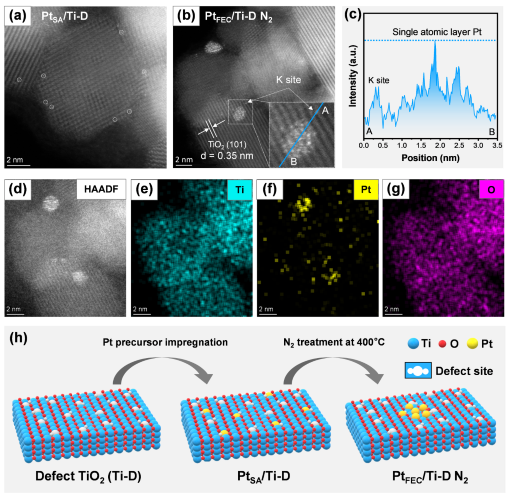
图2.(a)PtSA/Ti-D和(b)PtFEC/Ti-D N2的HRTEM图、(c)Pt团簇线轮廓图及(d-g)EDS元素映射图和(h)PtFEC/Ti-D N2催化剂合成过程示意图。
为了探究N2处理对Pt物种局域环境和配位结构的影响,采用XAFS技术对Pt L3边进行了详细分析。XANES的结果显示(图3a),PtFEC/Ti-D N2的白线峰强度低于PtSA/Ti-D,这表明在N2处理过程中,催化剂上Pt物种的氧化状态有所降低。通过与Pt箔和PtO2的白线峰强度进行对比,进一步确认了PtSA/Ti-D和PtFEC/Ti-D N2上的Pt物种主要处于氧化状态,而N2处理降低了Pt的氧化程度。这一结果可能与N2处理过程中缺陷结构的重组和电子转移有关,电子从载体表面转移到Pt物种上,从而实现了部分Pt氧化物的还原。为了更全面地阐明Pt物种的配位结构,采用EXAFS进行了分析。如图3b所示,PtSA/Ti-D在大约1.6 Å-1处显示了对应于Pt-O配位的主峰,这反映了Pt与其周围的氧原子的强相互作用。此外,在大约2.8 Å-1和3.3 Å-1处观察到的两个较弱峰,归因于Pt-Ti配位,表明Pt与载体表面Ti原子之间存在一定的相互作用。
为了进一步研究Pt原子中心周围邻近原子的特征,利用小波变换(WT)对XAFS光谱进行了分析。如图3b所示,PtSA/Ti-D的最大贡献度出现在R≈1.4-1.8 Å和k≈4.0 Å-1范围内,这归因于第一壳层中的Pt-O散射路径。而在R≈3.0−3.5 Å和k≈4.0 Å-1范围内,较小的强度最大值则对应于Pt-Ti散射路径的贡献。相比之下,PtFEC/Ti-D N2在第一壳层中主要显示Pt-O散射路径,同时出现了新的Pt−Pt散射路径(R≈2.6 Å,k≈9 Å-1)(图3d)。这一结果进一步验证了N2处理诱导了Pt物种从孤立原子向部分团聚状态的转变。
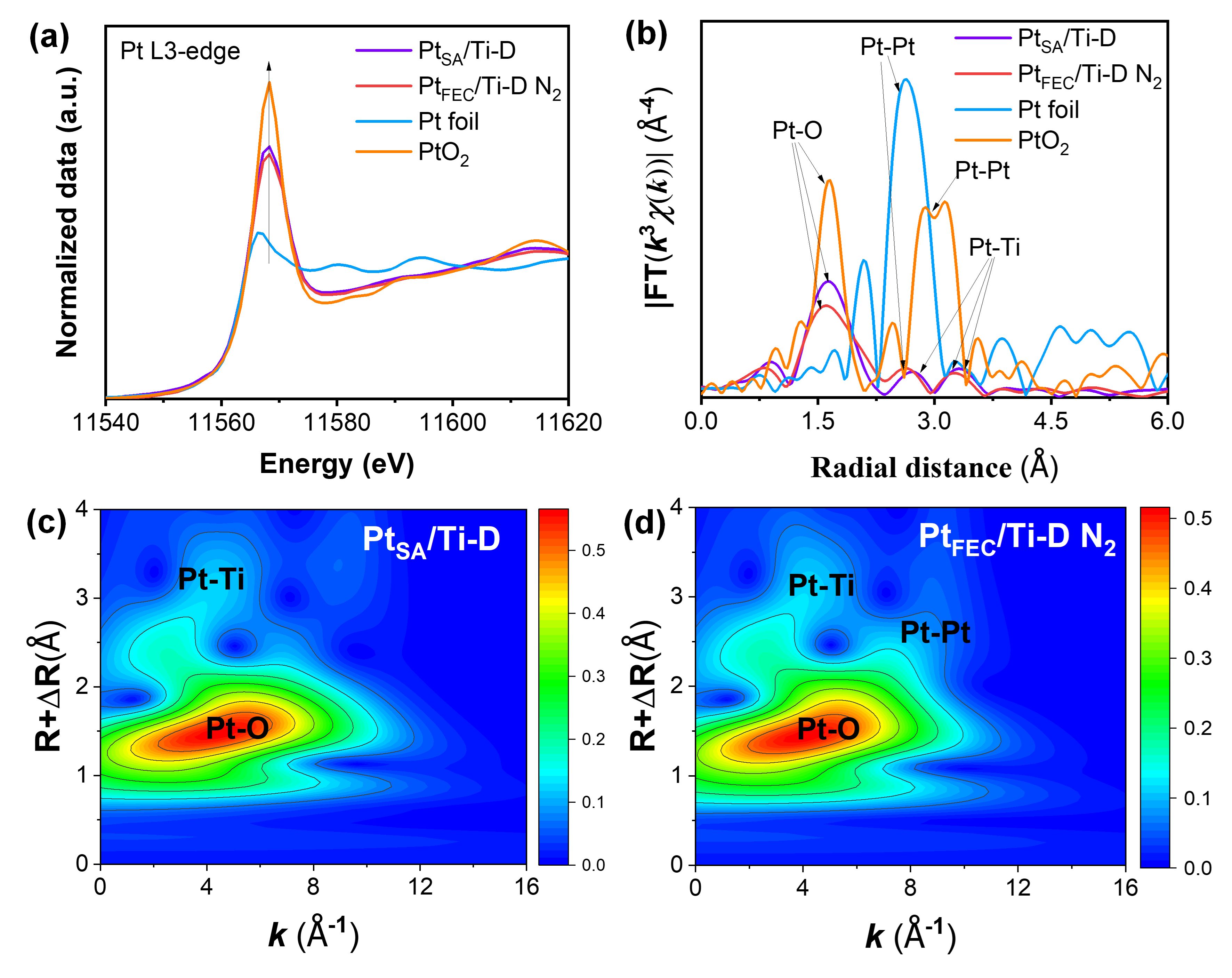
图3. PtSA/Ti-D、PtFEC/Ti-D N2、Pt箔和PtO2的(a)Pt L3边归一化XANES光谱和(b)Pt L3边缘k3加权傅里叶变换EXAFS光谱;(c和d)PtSA/Ti-D、PtFEC/Ti-D N2的Pt-L3边缘k3加权EXAFS信号的小波变换图。
3. 催化剂上CO的吸附行为
采用In situ DRIFTS研究了PtSA/Ti-D和PtFEC/Ti-D N2负载的Pt物种对CO的吸附行为。在50、100、150和200 ℃采集了两种样品的CO原位漂移光谱。如图4a所示,PtSA/Ti-D在50 ℃时除CO气相峰外,未观察到其他CO吸附峰;随着温度的升高,CO吸附峰出现。2072 cm−1和2057 cm−1处的CO峰归属于Ptδ+-CO,而2003 cm−1和1960 cm−1处的CO吸附峰归属于Pt缺陷位点上的CO吸附(PtDδ+-CO)。在50 ℃时,由于PtSA/Ti-D(CN = 6.1±0.3)上存在的Pt物种上的饱和Pt-O配位阻碍了CO吸附,因此没有出现CO吸附峰。然而,在较高温度(100、150和200 ℃)下,Pt-O键的断裂为CO的吸附提供了新的位置。吸附CO后,催化剂上形成Pt-CO-O-Ti桥型中间体,在2003 cm−1和1960 cm−1处观察到相应的CO吸附峰(PtDδ+-CO)。N2处理后,PtSA/Ti-D表面的Pt单原子聚集,导致Pt物种的化学结构发生显著变化。图4b显示了不同温度下PtFEC/Ti-D N2上CO的原位漂移光谱。与PtSA/Ti-D相比,PtFEC/Ti-D N2在50 ℃时,在2076 cm-1和2057 cm−1处观察到显著的CO吸附峰,证实了Pt物种的显著结构差异。随着温度的升高,CO吸附峰的信号强度显著增加,在2003 cm−1和1960 cm−1处出现了对应于PtDδ+-CO的吸收峰。PtFEC/Ti-D N2在50 ℃时的CO吸附峰是由Pt物种的不饱和配位引起的(CN = 5.4±0.4)。PtFEC/Ti-D N2具有优异的低温吸附性能。
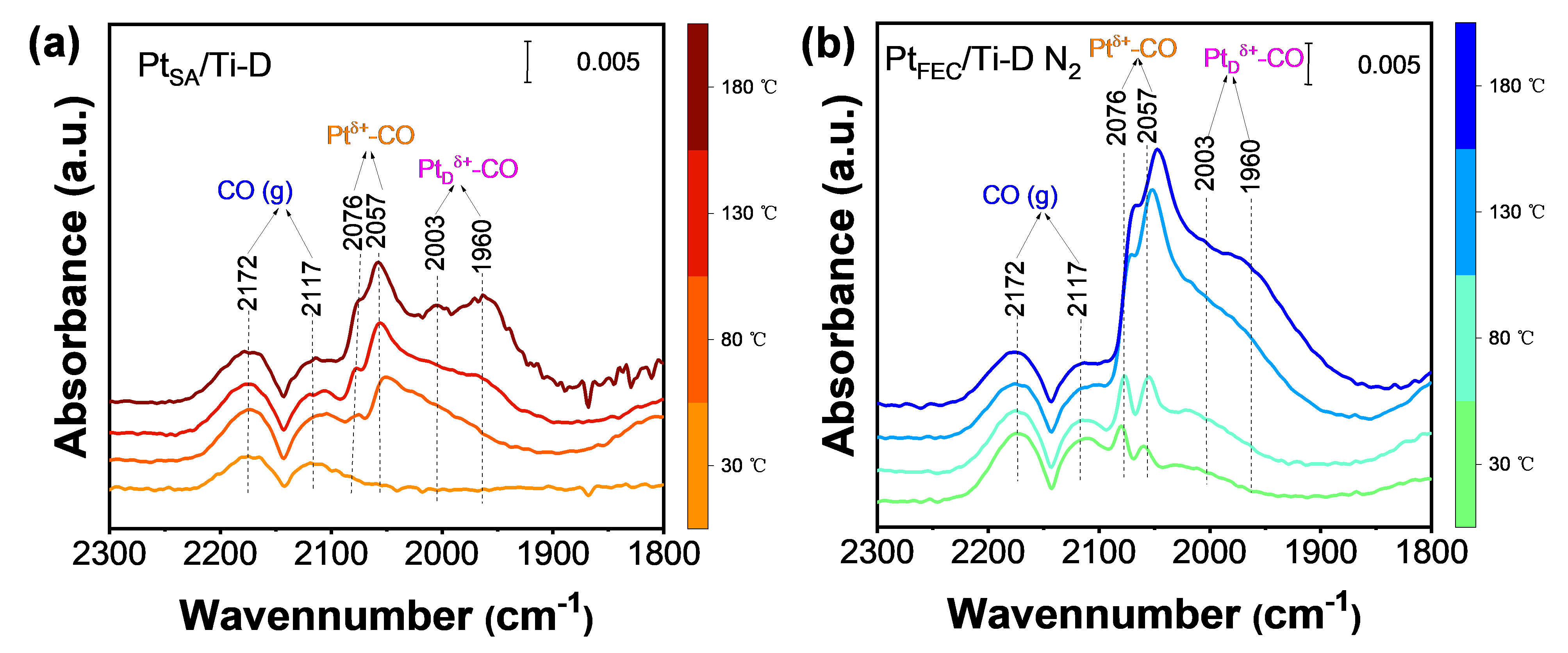
图4.(a)PtSA/Ti-D和(b)PtFEC/T-D N2暴露在1 vol % CO和50-200 °C下的In situ DRIFTS光谱。
采用密度泛函理论(DFT)研究了CO和O2在PtSA/Ti-D和PtFEC/Ti-D N2上的吸附性能。基于结构表征结果,建立了PtSA/Ti-D单原子催化剂和PtFEC/Ti-D全暴露团簇催化剂的模型。CO和O2在催化剂表面的吸附行为对两种CO氧化机理至关重要。CO和CO+O2在PtSA/Ti-D表面的吸附构型如图5a和图5b所示。当PtSA/Ti-D暴露在CO+O2气氛中时,CO通过其碳原子与Pt表面形成强σ键,优先吸附在Pt位。CO一旦吸附在接近饱和配位的Pt单原子上,O2就很难被吸附。Pt原子附近的缺陷结构是O2的理想吸附位置。图5c和图5d显示了CO和CO+O2在PtFEC/Ti-D N2表面的吸附构型。与PtSA/Ti-D相比,PtFEC/Ti-D N2上全暴露Pt团簇为CO和O2提供了更多的吸附位点。O2可以吸附在缺陷位点,也可以吸附在Pt位点。
如图5e所示,系统研究了CO和O2在PtSA/Ti-D和PtFEC/Ti-D N2表面的吸附能。在PtSA/Ti-D表面,单个Pt原子对CO的吸附能为-1.71 eV,明显低于PtFEC/Ti-D N2全暴露的Pt团簇对CO的吸附能-2.75 eV。值得注意的是,当PtSA/Ti-D和PtFEC/Ti-D N2都暴露在CO+O2气氛中时,两种催化剂之间的吸附能差异变得更加明显。PtSA/Ti-D对CO+O2的吸附能为-2.84 eV,而PtFEC/Ti-D N2对CO+O2的吸附能为-4.79 eV,表明Pt-Pt金属配位的Pt团簇比孤立的单个Pt原子具有更高的吸附能力。

图5. PtSA/Ti-D上(a)CO和(b)CO+O2的吸附构型,PtFEC/Ti-D N2上(c)CO和(d)CO+O2的吸附构型,(e)两个催化剂上CO和CO+O2的吸附能量。
总结与展望
综上所述,通过对Pt单原子缺陷催化剂进行N2处理,成功合成了全暴露Pt簇催化剂。经过N2处理的全暴露Pt簇催化剂在Pt负载量为0.1 wt.%下表现出优异的CO氧化性能,在130 ℃下即可实现CO完全氧化,相比未处理的Pt单原子缺陷催化剂降低70 ℃。综合表征表明,N2处理促进了缺陷结构的重构和演化,促进了Pt单原子聚集成全暴露的Pt团簇。全暴露Pt团簇在第一配位壳中表现出Pt-O配位减少,促进了Pt-Pt金属配位的形成。这种独特的配位环境不仅改善了CO的吸附,还增强了O2的活化,从而产生了优异的催化性能。本研究为设计和合成高效的低Pt催化剂提供了一种有前景的策略,为其在环境保护和大规模CO氧化过程中的工业应用提供了新的思路。
原文信息
相关成果以“N2 treatment triggered self-reorganization into fully exposed platinum cluster catalysts for efficient low-temperature CO oxidation”为题发表在Green Energy & Environment期刊,通讯作者为中国科学院过程工程研究所刘霄龙研究员。

扫码获取全文
https://doi.org/10.1016/j.gee.2025.04.005
撰稿:原文作者
编辑:GEE编辑部

转载本文请联系原作者获取授权,同时请注明本文来自何宏艳科学网博客。
链接地址:https://wap.sciencenet.cn/blog-3393673-1486927.html?mobile=1
收藏