博文
使用DESeq2进行转录组原始count标准化和差异分析
|
转录组测序完成后,一般我们会获得一个原始 read count表达矩阵,其中行是基因,列是样品。常用的差异分析工具包括limma、edgeR和DESeq2。DESeq2在测序领域使用最为广泛(google scholar引用高达43284次,edgeR为28076次)。小编今天给大家介绍下我们的在线DESeq2差异分析模块,小伙伴们可以零代码进行GEO数据库表达矩阵的挖掘,后续再利用我们平台的各种绘图模块出图,大大加速了我们的科学研究。
1,打开绘图页面
首先,使用浏览器(推荐chrome或者edge)打开DESeq2差异分析页面。左侧为常见作图导航,中间为数据输入框和可选参数,右侧为描述和结果示例。也可以在搜索框中搜索deseq2,找到分析页面。

图1,DESeq2分析页面
2,示例数据
点击右侧“示例数据”链接下载excel格式的示例数据。
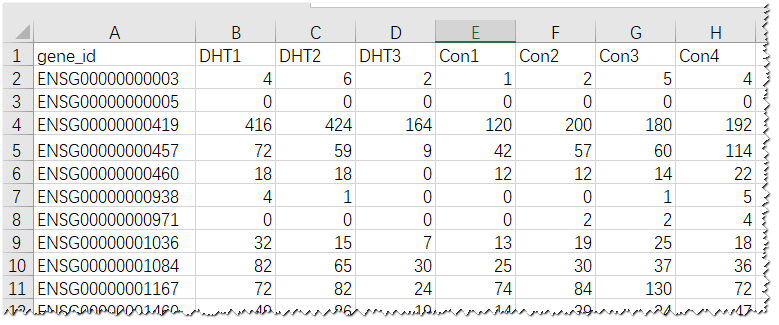 图2. Raw count输入数据示例
图2. Raw count输入数据示例
示例raw count矩阵包括8列:
第1列是基因id,必需唯一;
第2-4列是DHT组3个样品;
第5-8列是Control组4个样品。
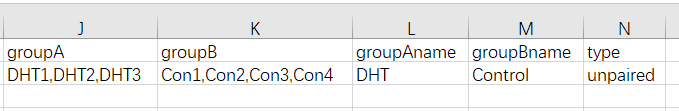
图3. 比较方案输入数据示例
示例分组比较方案包括5列:
第1列是实验组的样品列表,共3个,英文逗号分隔
第2列是对照组的样品列表,共4个,英文逗号分隔
第3列是实验组组名
第4列是对照组组名
第5列是样品配对与否,unpaired表示非配对,paired表示配对。例如从1号、2号、3号共三个患者身上分别取癌症样品cancer1,cancer2,cancer3,对照样品norm1,norm2,norm3,那么paired配对比较时,顺序不能变,即:1号患者的cancer1,对应1号患者的norm1;2号患者的cancer2对应2号患者的norm2;3号患者的cancer3对应3号患者的norm3。而非配对比较(unpaired)则不考虑这种对应关系。
3,粘贴示例数据
拷贝示例数据中A-H列的raw count数据,粘贴到矩阵输入框。
拷贝J-N列的比较信息,粘贴到比较输入框。
注意:不是拷贝excel文件,是拷贝excel文件里边的数据。另外粘贴到输入框后,格式乱了没关系,只要在excel中是整齐的就行。同时数据矩阵中不能有空的单元格,中文字符等。
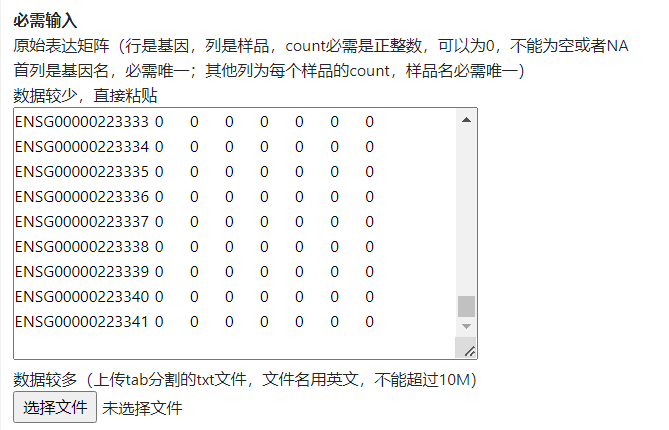
图4. 必需输入-raw count矩阵

图5. 必需输入-比较方案
4,修改参数,并提交
我们设置了原始count数之和的过滤最小过滤参数,默认不过滤。

图6.过滤参数
5,提交获得结果
粘贴好输入数据,调整好参数后,点击提交按钮,约60秒钟后,会在页面右侧出现结果下载链接。下载并解压后,可以使用excel打开并利用筛选功能根据fold change和p值筛选差异表达基因,绘制热图,火山图,进行功能富集分析。

图7.结果下载
结果说明
由于我们未对低表达值进行过滤,因此,输入矩阵是多少个基因,分析结果也是多少个基因。
第1个sheet是表达谱,包括原始count和标准化count
第2+个sheet是差异总表。
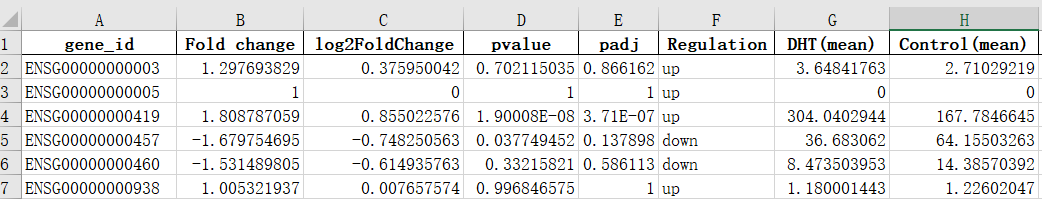 图8. 差异结果
图8. 差异结果
Fold change:两组间的倍数变化。正表示上调,负表示下调
Log2FoldChange:log2转化的倍数变化。正表示上调,负表示下调
Pvalue:p值
Padj:校正的p值
Regulation:上调或下调
*(mean):各组的标准化表达均值
不同平台相互印证测试
除了阅读DESeq2官方文档并使用官方代码外,我们还使用相同的raw count矩阵测试了其他4个第三方平台:DEApp,基迪奥,欧易,sangerbox。在这5个平台中,微生信的结果和sangerbox完全一样,而与其他3个平台的结果均略有差异(见图9)。图中所示785个基因是4种平台共有的,约占90%以上。

图9. 4种平台上调结果比较
究其原因,可能主要是内部filter过滤参数不一致造成的。在DESeq2官方文档中:While it is not necessary to pre-filter low count genes before running the DESeq2 functions, there are two reasons which make pre-filtering useful: by removing rows in which there are very few reads, we reduce the memory size of the dds data object, and we increase the speed of the transformation and testing functions within DESeq2. It can also improve visualizations, as features with no information for differential expression are not plotted.
意思就是过滤低表达量的基因不是必需的,过滤的用途包括:1,减少dds对象内存占用,提高计算速度;2,改善可视化。可以使用所有样品的均值过滤,也可以使用组内样品的均值进行过滤,在不确定过滤参数的情况下会出现结果差异的现象。
虽然我们参考的都是官方文档,但是由于大家对官方文档的具体细节处理、具体参数使用等存在差异,因此导致各个平台的结果略有差异,但是总体相差较小,基本不影响结果。
微生信助力高分文章,用户58000+,引用750+
https://wap.sciencenet.cn/blog-707141-1363668.html
上一篇:实验送样、数据分析样品、组名命名规范、
下一篇:带临床数据的热图 -- 给样品添加TNM分期、年龄、性别、riskscore等信息