博文
lncRNA研究生物信息学工具推荐
||
lncRNA研究生物信息学工具推荐
长链非编码RNA (lncRNAs)通常被定义为长度超过200个核苷酸(nt)的非蛋白质编码RNA。这些分子最初被认为是从垃圾DNA区域转录而来的噪音。 然而,二十多年的研究表明,许多lncRNA通过与其他大分子(包括DNA、RNA和蛋白质)的结合,在微调信号通路方面具有独特的功能。鉴于以蛋白质为中心的研究一直无法完全阐明许多生物现象和疾病背后的复杂信号通路和机制,人们非常希望包括lncRNA在内的非编码RNA能够为更好地理解这些过程提供见解。此外,虽然大多数哺乳动物基因组被转录为RNA,但仅鉴定了多达20,000个蛋白质编码基因,随着基因检测和注释技术的成熟,这一数字已被下调。相比之下,lncRNA的数量超过蛋白质编码基因(人类蛋白质编码基因为19,379个,lncRNA基因为19,933个,于2023年2月4日查阅https://www.gencodegenes.org/human/stats_42.html),并且估计正在迅速增加(根据NONCODE数据库,96,411个人类lncRNA基因,于2023年2月4日查阅http://www.noncode.org)。
然而,由于许多这些lncRNA可能没有功能,进一步的鉴定和注释lncRNA是生物信息学家的重要任务,因为它为实验科学家和临床医生进行的功能和机制实验提供了最相关的靶标。事实上,尽管lncRNA的功能重要性越来越被认识到,但在缺乏详细机制数据的情况下,特别是在临床应用背景下,如lncRNA作为各种疾病治疗药物的潜在用途,推动lncRNA领域向前发展是具有挑战性的。尽管正在进行临床试验研究lncRNA与疾病之间的关(https: //clinicaltrials.gov/ct2/results? cond=&term=long+non-coding+rna),lncRNA的研究落后于其他非编码RNA,如microRNAs (miRNAs)。由于lncRNA的许多未知方面,很难有效地靶向实验工作。因此,优先考虑lncRNA的功能和机制研究是至关重要的。为此,生物信息学工具可以在鉴定、注释和预测lncRNA的潜在功能方面发挥重要作用,从而使实验更加集中和有针对性地阐明每个lncRNA的作用机制。虽然lncRNA领域正在认识到生物信息学工具应用于lncRNA,但目前可用的工具数量庞大。因此,在不断增长的生物信息学工具集合中导航可能很困难,特别是对于计算机编程技能有限的实验科学家和临床医生。为了驾驭这一系列复杂的生物信息学工具,在这里,Rebecca等人将重点放在认为对鉴定、注释和预测lncRNA潜在功能最有用的工具上。其目标是减轻与寻找最合适的lncRNA研究生物信息学工具相关的困难。此外,该建议也适用于分析蛋白质编码RNA。如需希望修改建议的管道,需要注意其他可供选择的工具。
从RNA-seq数据中发现lncRNA的生物信息学工具
由于RNA-seq是目前最广泛使用的分析蛋白质编码和lncRNA基因表达的方法,许多lncRNA已被发现使用这种类型的数据,以及它们的组织特异性背景和条件,其中它们在应激或疾病中失调。然而,使用生物信息学工具分析RNA-seq数据对实验科学家和临床医生来说可能是一个挑战,因为大多数这些工具缺乏图形用户界面(GUI),只能作为命令行工具使用。 虽然市面上有一些用户友好的一体化工具(https://en.wikipedia.org/wiki/List_of_RNASeq_bioinformatics_tools)。针对非专业生物信息学家的一个免费选项是Galaxy,这是一个旨在提高可用性的平台。 Galaxy包含大量端到端工作流,包括RNA-seq处理和分析。然而,大量可用的工具可能是压倒性的,而最终结果因每个生物信息学工具选择的参数而有很大差异,这一事实进一步加剧了这一情况。对于感兴趣的读者,有几篇优秀的文章比较了每种生物信息学工具的性能及其用于分析RNA-seq数据的参数。基于在分析RNA-seq数据和类似分析方面的丰富经验,作者们提出了以下计算方法来进行大量RNA-seq分析(图1)。 首先,FASTQ文件必须进行预处理,包括质量控制、适配器修剪、质量过滤和读取修剪。这些任务可以通过使用默认设置运行生物信息学工具fastp轻松完成。此工具可从GitHub (https://github.com/OpenGene/fastp)获取,其中还提供了安装和使用的详细协议。另一种常用的RNA-seq 读取数质量控制工具是FASTQC,尽管它通常比fastp慢。
在预处理之后,修剪的读数需要被映射到参考基因组。为此,STAR、HISAT2和TopHat2是一种流行的快速读取的校准器。STAR可通过GitHub (https://github.com/alexdobin/STAR)获取,里面有安装和使用的详细协议。请注意,通常建议使用至少有16GB随机存取存储器(RAM)的计算机进行映射。此外,建议提供从Ensembl数据库中获得的目标物种的基因组构建和GTF文件(http://www.ensembl.org/info/data/ftp/index.html/)。在运行首选比对工具之前,有必要为相同的物种和基因组构建生成参考基因组的索引。此外,应使用最新版本的相关GTF文件,特别是考虑到已鉴定的lncRNA基因数量不断增加。将测序读数映射到参考基因组后,需要将读数归一化,以便在不同条件(例如,患病与健康)之间进行比较,从而能够识别差异表达的基因。为此,建议使用R包edgeR,以及另一个R包GenomicFeatures为规范化结果追加注释。这两个工具都可以从Bioconductor项目(https: //www.bioconductor.org/)获取。对于生成图(例如火山和小提琴图),建议使用R包ggplot2。作为参考,作者们概述了四种主要的表达规范化类型:1)每千碱基转录片段每百万映射读取(FPKM);2)每百万读数(CPM);3)每千碱基的转录本每百万映射读数(RPKM);以及4)每百万转录本(TPM)。关于RNA-seq表达数据规范化的争论正在进行中,尽管更喜欢CPM值,以避免与备选剪接和同工异构体长度相关的潜在问题。最后,对于涉及特定条件或研究较少的疾病RNA-seq分析,人们考虑转录本的从头组装。这可能会揭示出对这些条件具有高度特异性且尚未包含在当前注释中的新lncRNA。对于从头组装,流行的工具包括Oases (https://github.com/dzerbino/oases),SOAPdenovo-Trans (https://sourceforge.net/projects/soapdenovotrans/),StringTie (https://ccb.jhu.edu/software/stringtie),Trinity (https://github.com/trinityrnaseq/trinityrnaseq)。由于每个工具的性能可能会因基因组的注释程度而有很大差异,因此读者可以参考最近的文章,这些文章对不同的从头组装工具的性能进行了基准测试。尽管专注于大量RNA-seq数据,单细胞RNA-seq (scRNA-seq)正在成为分析表达变化的主流分析方法。然而,与蛋白质编码基因相比,许多lncRNA的表达水平较低,即使在细胞培养的RNA-seq数据中也是如此。此外,只有一半的lncRNA具有poly-A尾巴。对于大量RNA-seq,这个问题是通过执行核糖体RNA (rRNA)耗尽的RNA-seq来捕获各种RNA物种,除了rRNA。然而,scRNA-seq的情况并非如此,因为大多数scRNA-seq文库制备的流行协议是基于使用oligo dT引物来扩增具有poly-A尾巴的RNA物种。由于这些原因,没有推荐从scRNA-seq数据中鉴定lncRNA的首选工具。
功能预测的生物信息学工具
推断已鉴定lncRNA潜在功能的最常用方法是检查共表达蛋白编码基因的功能类别。这可以很容易地通过向在线生物信息学工具提供差异表达的蛋白质编码基因列表来完成,例如DAVID (https://david.ncifcrf.gov)。该工具返回丰富的基因本体(GO)术语,BioCarta和KEGG通路。例如,它对于获得目标lncRNA在操纵(即功能的获得/丧失)时受影响的信号通路的概述是有价值的。对于喜欢基于R的解决方案的用户,gprofiler2也是确定功能富集程度的好选择,其结果与DAVID相似。此外,如果你想可视化信号传导通路,一个选择是在线生物信息学工具NetworkAnalyst (https://www.networkanalyst.ca/NetworkAnalyst/),它比更全面的Cytoscape工具更容易使用。目前,lncRNA领域的共识是lncRNA通过与其他大分子(即DNA、RNA和/或蛋白质)结合来发挥其功能。由于基于抗体的下拉测定法在蛋白质研究中已经很好地建立起来,因此可以使用几种基于下拉测定法的测序数据(例如,染色质免疫沉淀测序(ChIP-seq), RNA免疫沉淀测序(RIP-seq))。 方便的是,这些数据被收集起来,并以数据库和基因组查看器的形式提供。例如,在线生物信息学工具UCSC基因组浏览器(https://genome.ucsc.edu),包含ChIP-seq, RIP-seq,预测miRNA结合位点等结合数据。因此,在基因组观察器上直观地检查蛋白质和miRNA的潜在结合位点是可行的,这可能有助于确定lncRNA是作为蛋白质(例如,RNAbinding蛋白)还是miRNA海绵发挥作用。最后,RNA是高度动态分子,不仅仅以线性形式存在。相反,它们折叠形成三维结构,这对于它们与其他大分子的结合能力很重要。当使用RNA序列作为输入时,有几个有用的生物信息学工具可用于预测大分子的结合。在这些工具中,推荐在线生物信息学工具RNALigands (http://rnaligands.ccbr.utoronto.ca)。特别有趣的是,该工具以RNA序列作为输入,并返回在蛋白质数据库(PDB)和RBIND数据库中注册的类似RNA基序以及结合的小配体。这些预测信息有助于设计实验来寻找潜在的lncRNA功能抑制剂。如果候选lncRNA在疾病的进展中发挥明确的作用,这将促进小分子药物的开发。此外,Rfam数据库 (https://rfam.org)是包括lncRNA的RNA分子共识二级结构的宝贵信息来源。
lncRNA数据库
鉴定和表征lncRNA是推进lncRNA研究领域的重要任务。同样重要的是传播有关lncRNA的已知信息,以便与更广泛的研究界分享所获得的知识。为此,数据库是必不可少的工具。使用最广泛的lncRNA数据库是NONCODE (http://www.noncode.org),该数据库包括人类、小鼠、大鼠、鸡、斑马鱼和蠕虫等16个物种的所有类型的ncRNA(除了核糖体RNA(rRNA)和转运RNA (tRNA))。此外,LncBook 2.0 (https://ngdc.cncb.ac.cn/lncbook/)是另一个提供lncRNA综合注释的数据库。除了这些lncRNA数据库,RNAcentral (https://rnacentral.org)存储了来自不同物种的ncRNA的序列信息,而GeneCards (https://www.genecards.org)整合了来自150个网络来源的人类基因(包括lncRNA)的基因组学、转录组学、蛋白质组学、遗传学、临床和功能信息(例如,亚细胞定位)。这些数据库对于获得目标lncRNA的进一步信息是有用的。
讨论
图1中突出显示的生物信息学工具是根据已发表研究选择的,并通过lncRNA的生物实验进一步验证。值得注意的是,所选择的工具尚未与其他类似工具(例如,用于读取修剪的Cutadapt 、 FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)、prinseq和Trimmomatic;用于转录组组装的HISAT2、Kallisto、TopHat、Rnnotator和Trinity;用于差异表达分析的DEseq2和limma;用于GO分析的clusterProfiler、fgsea和GSEA)进行比较。因此,建议读者查阅对这些生物信息学工具进行基准测试的研究,以及优秀的综述文章,以便对推荐的管道进行潜在的修改。此外,作者们还提供了一份生物信息学工具的非详尽清单,发现这些工具在鉴定和表征lncRNA方面很有用。
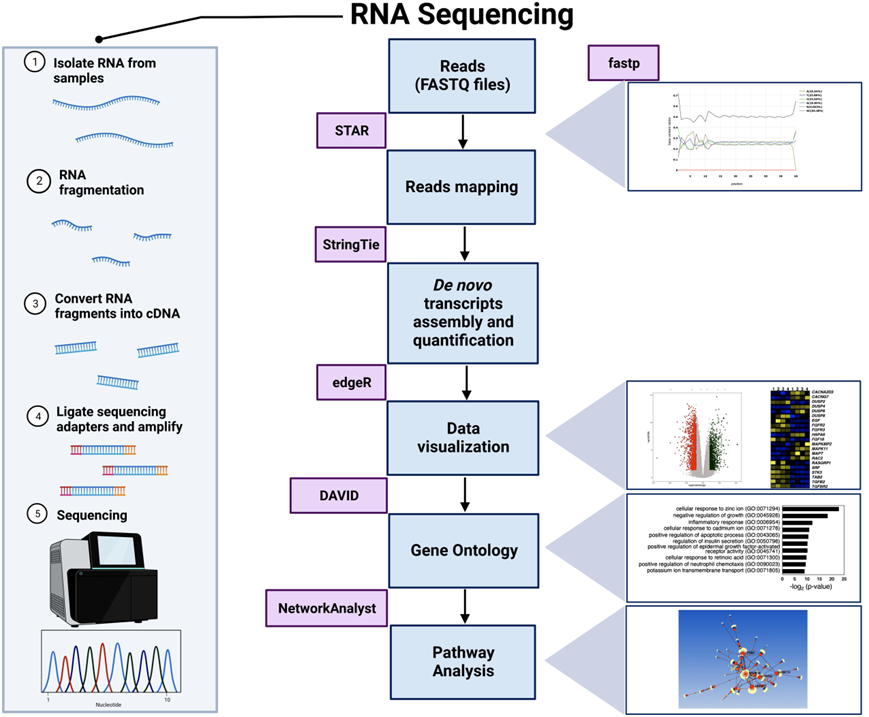
图1 RNA-seq数据分析建议。原始RNA-seq数据(FASTQ文件)可以使用fast程序进行预处理。然后可以使用STAR程序将修剪后的测序读数映射到参考基因组。对于从头组装转录本,可以使用StringTie程序。将读数定位到参考基因组后,可以使用R包edgeR识别差异表达基因。然后可以使用热图和火山图将这些可视化,这些热图和火山图是通过其他R包(如ggplot2)生成的。为了获得差异表达基因的概述,DAVID在线工具可用于识别富集的基因本体(GO)术语。最后,为了进一步可视化差异表达基因,NetworkAnalyst在线工具可能是有益的
目前还没有lncRNA被开发成一种用于晚期临床的治疗工具。鉴于lncRNA的数量和多样性,迫切需要进一步的功能和机制研究,以了解每种lncRNA在疾病背景下的作用。因此,对每个lncRNA进行基本信息注释是至关重要的,这些信息包括(但不限于)基因组位置、同工型(转录本)数量、编码潜力(如微肽)和结合伙伴。此外,每个lncRNA的特性都应该通过实验来证明。为此,建议进行以下实验:1)5/3 端互补DNA末端快速扩增(RACE)和Northern blotting进行异构体鉴定;2)体外转录/翻译实验,确认蛋白编码能力;3)荧光原位杂交(FISH)和逆转录聚合酶链反应(RT-PCR),利用从细胞核和细胞质部分分离的总RNA进行亚细胞定位;4)增益/功能损失实验;5) RNA下拉,质谱鉴定蛋白结合伙伴,通过RNA免疫沉淀确认结合,然后利用引物对靶向lncRNA进行RT-PCR (RIP-PCR)。总之,上述建议将有助于通过生物学实验对lncRNA候选者进行功能和机制表征的优先排序。这将在每个信号通路中定位lncRNA,并将增强我们对它们的失调如何在机制上促进疾病进展的理解。
参考文献
[1] Distefano Rebecca, Ilieva Mirolyuba, Rennie Sarah, Uchida Shizuka, Recommendations for Bioinformatic Tools in lncRNA Research, Current Bioinformatics 2024; 19 (1), doi:10.2174/1574893618666230707103956
以往推荐如下:
5. EMT标记物数据库:EMTome
8. RNA与疾病关系数据库:RNADisease v4.0
9. RNA修饰关联的读出、擦除、写入蛋白靶标数据库:RM2Target
13. 利用药物转录组图谱探索中药药理活性成分平台:ITCM
19. 基因组、药物基因组和免疫基因组水平基因集癌症分析平台:GSCA
22. 研究资源识别门户:RRID
24. HMDD 4.0:miRNA-疾病实验验证关系数据库
25. LncRNADisease v3.0:lncRNA-疾病关系数据库更新版
26. ncRNADrug:与耐药和药物靶向相关的实验验证和预测ncRNA
28. RMBase v3.0:RNA修饰的景观、机制和功能
29. CancerProteome:破译癌症中蛋白质组景观资源
30. CROST:空间转录组综合数据库

https://wap.sciencenet.cn/blog-571917-1424909.html
上一篇:TargetRNA3:用机器学习预测原核RNA调控靶标
下一篇:lncHUB2:汇总和推断人类和小鼠lncRNA知识