ВЉЮФ
ЙЙНЈдКЫЩњЮяЛљвђзщНјЛЏЪїЃПЪдЪдEasyCGTreeЃЌШчДЫМђЕЅЃЁЃЁЃЁЁОКЌЪгЦЕНЬГЬЁП
|
ЙЙНЈдКЫЩњЮяЛљвђзщНјЛЏЪїЃПЪдЪдEasyCGTreeЃЌШчДЫМђЕЅЃЁЃЁЃЁЁОКЌЪгЦЕНЬГЬЁП
в§бдЃК2023Фъ10дТ14ШеЃЌЩњЮяаХЯЂбЇжЊУћЦкПЏЁЖBMC BioinformaticsЁЗЩЯЯпСЫЬтЮЊЁЖEasyCGTree: a pipeline for prokaryotic phylogenomic analysis based on core gene setsЁЗЕФЮФеТЁЃНщЩмСЫвЛПюУћЮЊEasyCGTreeЕФШэМўЃЌПЩвдЪЕЯжДгЛљвђзщЕААзжЪађСаЕНЛљвђзщНјЛЏЪїЕФвЛВНВйзїЃЌМђЛЏСЫДЋЭГЕФЭЌдДЛљвђМьЫїЁЂађСаБШЖдЁЂДЎСЊЕШвЛЯЕСаЗБЫіСїГЬЁЃ
ШэМўЯТдиЕижЗЃК
https://github.com/zdf1987/EasyCGTree4
https://gitee.com/zdf1987/EasyCGTree4
ЮФеТСДНгЃКhttp://dx.doi.org/10.1186/s12859-023-05527-2
ЫцзХЛљвђзщЪБДњЕФЕНРДЃЌЛљгкЛљвђзщНјааЯЕЭГЗЂг§ЗжЮівбОБфЕУдНРДдНЦеБщЁЃгаСНжжЗжЮіВпТдзюЮЊГЃМћЃЌЗжБ№БЛГЦЮЊSupermatrixЃЈSMЃЉКЭSupertreeЃЈSTЃЉЁЃSMЪЧНЋБШЖдКУЕФИїИіЛљвђађСаДЎСЊЦ№РДаЮГЩвЛЬѕЁАГЌМЖађСаЁБРДНјааНјЛЏЪїЕФЙЙНЈЃЌЖјSTЪЧНЋИїИіЛљвђЗжБ№ЙЙНЈЛљвђЪїЃЈPhylomeЃЉЃЌШЛКѓЪЙгУЬиЖЈЕФЫуЗЈНЋЛљвђЪїећКЯГЩвЛИіНјЛЏЪїЁЃ
дкдКЫЩњЮяжаЃЌSMЕФНЈЪїВпТдЪЧзюЮЊГЃгУЕФЁЃЕЋЪЧЃЌећИіСїГЬЩцМАШэМўжкЖрЃЌЯрЙиШэМўЕФАВзАЁЂЪ§ОнЕФзМБИЁЂИёЪНДІРэЕШВйзїЖМашвЊвЛЖЈЕФзЈвЕММФмКЭОбщЁЃвђДЫЃЌЛљвђзщЯЕЭГЗЂг§ЗжЮіЖдгкГѕбЇепРДНВЃЌММЪѕФбЖШНЯИпЁЃEasyCGTreeБужТСІгкЬсЙЉвЛжжМђЕЅЕФЗНЗЈЁЃ

EasyCGTreeЪЙгУHMMERНјааЭЌдДЛљвђМьЫїЃЌMUSCLEКЭClustal OMEGAНјааађСаБШЖдЃЌtrimAIНјааБЃЪиЧјгђЩИбЁЃЌFastTreeКЭIQ-TREEНјааНјЛЏЪїЕФЙЙНЈЃЛПЩвдЪЕЯжSMЁЂSTКЭconsensus treeЃЈвЛжТадЪїЃЌSMЕФСэвЛжжаЮЪНЃЉШ§жжНЈЪїВпТдЁЃдкБОЮФЗЂБэжЎЧАЃЌEasyCGTreeвбОдкаЁЗЖЮЇФкЪдгУЃЌВЂБЛЖрЦЊвбЗЂБэТлЮФв§гУЁЃгыЯжгаОпгаРрЫЦЙІФмЕФШэМўautoMLSTЁЂbcgTreeЁЂGToTreeКЭUBCGЯрБШЃЌEasyCGTreeжЇГжWindowsКЭLinuxЃЌВЛашвЊАВзАЕкШ§ЗНШэМўЃЈвбЬсЧАДђАќХфжУКУЃЉЃЌздДјbac120ЁЂar122ЁЂrp1КЭrp2ЕШЛљвђМЏЕФHMMЮФМўЃЛжЇГжЛљвђМЏHMMsРЉеЙЃЌВЂЧвЪЕЯжСЫSTВпТдНЈЪїЁЃжЛашвЊАВзАЛљБОЕФPerlгябдЛЗОГЃЌБуПЩвддЫааEasyCGTreeЁЃ

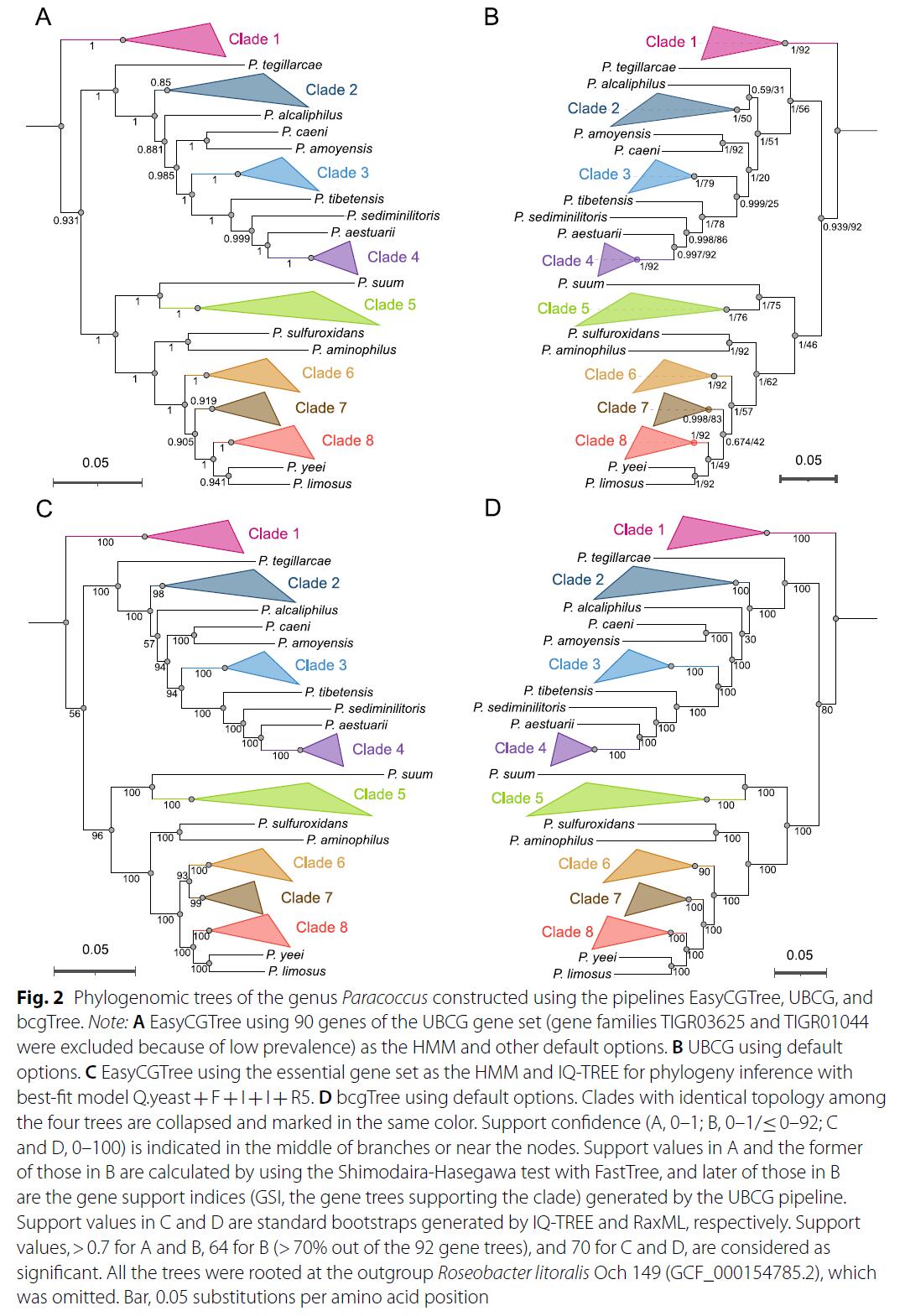
зїепвдParacoccusЪєЮЊР§ЃЌЙЙНЈСЫЖрИіНјЛЏЪїЃЌВЂгквбЗЂБэЕФbcgTreeКЭUBCGШэМўЕФНсЙћНјааБШНЯЁЃНсЙћЯдЪОЃЌдкЗНЗЈРрЫЦЕФЧщПіЯТEasyCGTreeПЩвдЙЙНЈгыbcgTreeКЭUBCGЭиЦЫНсЙЙИпЖШвЛжТЕФНјЛЏЪїЃЌЭЌЪБдЫаааЇТЪИќИпЃЌКФЪБИќЖЬЃЌвВИќМгЪЪКЯгУгкКЃСПЛљвђзщЪ§ОнЕФЗжЮіЁЃ
EasyCGTreeЕФЪЙгУвВЗЧГЃМђЕЅЁЃЪзЯШАВзАPerlгябдЃЌШЛКѓЯТдиШэМўАќКЭШэМўЬсЙЉЕФHMMЪ§ОнЃЌНтбЙКѓМДЭъГЩАВзАЁЃ
ЪЙгУЗНЗЈЃК
1ЁЂзМБИД§ЗжЮіОњжъЕФCDSАБЛљЫсађСа
ШЗБЃАќКЌађСаЕФЮФМўМа(Р§ШчmyGenome)дкEasyCGTreeЕФжїФПТМЯТ(Р§ШчLinuxЕФЁАЁ/Downloads/EasyCGTreeЁБ)ЃЛЁА/ЁБЖдгкWindowsгІИУЪЧЁА\ЁБ)ЃЌНЋЙЄзїФПТМИќИФЮЊEasyCGTreeЕФЙЄзїФПТМВЂдЫааЁЃ
МйЩшWindowsгУЛЇНЋШэМўЯТдиЕНЁАD:ЁБХЬЯТЕФЁАDownloadsЁБЮФМўМажаЁЃ
2ЁЂдЫааEasyCGTree
зЂвтЃКEasyCGTreeЪЧвЛИіУќСюааШэМўЃЌашвЊдкcmd.exe (Windows)ЛђTerminal (Linux)жадЫааЁЃУќСюаадЫааЕФЪОР§ШчЯТ(дкЭъГЩУПвЛааЪБАДЁАEnterЁБ)ЃК

LinuxгУЛЇЪЙгУЁАcdЁБУќСюНЋEasyCGTreeЕФЙЄзїФПТМИФЮЊEasyCGTreeЕФЙЄзїФПТМКѓЃЌПЩвдАДееЯрЭЌЕФЗНЪНдЫааEasyCGTreeЁЃ
ШЛКѓЃЌМДПЩЛёЕУЪЙгУФЌШЯВЮЪ§ЃЈbac120ЛљвђМЏЃЌtrimAlЕФЁАstrictЁБбЯНїЖШЩИбЁБЃЪиЧјЃЌFastTreeЙЙНЈЃЉЙЙНЈЕФNewickИёЪНЕФНјЛЏЪїЃЈвдАќКЌЛљвђзщЕФЮФМўМаЕФУћГЦУќУћЃЌР§ШчmyGenome.bac120.supermatrix.fasttree.treeЃЉКЭдЫааЦкМфЩњГЩЕФЮФМў/ЮФМўМаЁЃПЩвдЪЙгУFigTree, MEGA, iTOLЛђЦфЫћНјЛЏЪїВщПДЦїРДЯдЪОНјЛЏЪїЁЃ
гХЛЏНЈЪїЗНЗЈЃЌвВКмМђЕЅЃК
ЯыЪЙгУЦфЫћЛљвђМЏЃЌдкзюКѓвЛааУќСюЬэМгЁА -hmm ar122/rp1/rp2ЛђЦфЫћЁБМДПЩЃЈФЌШЯbac120ЃЉЁЃ
дЫааЬЋТ§ЃЌЯыдіМгЯпГЬЪ§ЃЌЬэМгЁА-thread Ъ§зжЁБ ЃЈФЌШЯ2ЃЉЁЃ
ЯыЪЙгУIQ-TREEНјааНјЛЏЪїЙЙНЈЃЌЬэМгЁА-tree_app iqtreeЁБ ЃЈФЌШЯfasttreeЃЉЁЃ
ЯыЙЙНЈsupertreeНјЛЏЪїЃЌЬэМгЁА-tree stЁАЃЈФЌШЯsmЃЉЁЃ
ЯыИФБфtrimAlЕФбЯНїГЬЖШЃЌЬэМгЁА-trim gappyout/strictplusЁАЃЈФЌШЯstrictЃЉЁЃ
ЯыаоИФFastTree/IQ-TREEЕФВЮЪ§ЃЌПЩдкЁАtree_app-options.txtЁБЮФМўжаНјааЁЃ
зюКѓЃЌеыЖддКЫЩњЮяЗжРрбЇЯрЙиЕФбаОПЃЌзїепЛЙЬсЙЉСЫДгЪ§ОнзМБИЕНЛёЕУНјЛЏЪїЯъЯИЕФВйзїЪгЦЕЁЃ
ЁОШчКЮХњСПЯТдиType strainЛљвђзщЪ§ОнЁП https://www.bilibili.com/video/BV14w41167FR/?share_source=copy_web&vd_source=e5f4fe144d94ec05eb13374f0dd44d7d
ЁОЪЙгУEasyCGTreeЙЙНЈдКЫЩњЮяЛљвђзщНјЛЏЪїЁП https://www.bilibili.com/video/BV1KN411t7pX/?share_source=copy_web&vd_source=e5f4fe144d94ec05eb13374f0dd44d7d
https://wap.sciencenet.cn/blog-548406-1406360.html
ШЋВПОЋбЁВЉЮФЕМЖС
- • Ь§ЕМЪІЕФЛАЃЌБЯвЕТлЮФД№БчетбљзМБИ
- • Accounts of Materials Research ЧАеАадЙлЕуЮФеТ(Viewpoint)ОЋбЁЃЈЮхЃЉ
- • змНБН№ 300 ЭђШ№ЪПЗЈРЩЃК2024 ФъЖШ ЁАЧАбиЕиЧђНБЁБ МДНЋНвЯў
- • ПЦбЇМвУЧРЇЛѓСЫ50ЖрФъЃЌвЛИіЪЅБАуЕФЛЏбЇжЎУежегкНтПЊСЫ
- • ЕТЙњTU IlmenauРзгТЕШзлЪіЃКгУзїЯШНјЕчЛЏбЇДЂФмВФСЯЕФЮЂФЩжЇГХНсЙЙЁЊзюаТНјеЙЁЂбаОПЬєеНКЭЮДРДЧАОА
- • ЩѓИхШЫЧПжЦзїепв§гУздМКЭХЖгЮФЯзЕФЬзТЗ