博文
氧化应激在神经退行性疾病中的作用:活性氧和抗氧化剂的预防研究进展
|
氧化应激在神经退行性疾病中的作用:活性氧和抗氧化剂的预防研究进展
原文作者:英国半岛医学与牙科学院分子医学研究小组Annwyne Houldsworth
摘要:神经障碍包括多种疾病,如阿尔茨海默病、运动神经元疾病和帕金森病,这些疾病影响寿命和生活质量,其发病机制与氧化应激有关。中枢神经系统的几种慢性神经退行性疾病有一些共同特征,例如氧化应激、炎症、突触功能障碍、蛋白质错误折叠和有缺陷的自噬作用。神经炎症可能涉及肥大细胞的激活,导致氧化应激,此外还有其他活性氧物质的来源。抗氧化剂可以有效地中和活性氧物质和自由基,减少氧化损伤。抗氧化基因,如锰超氧化物歧化酶(MnSOD)酶,可能会经历表观遗传变化,从而降低它们的表达,因此增加组织中的氧化应激。或者,DNA可以被自由基损伤所改变。这些基因的表观遗传景观的变化可能会改变抗氧化功能,并可能导致神经退行性疾病。自由基产生和抗氧化功能的不平衡增加了导致神经元细胞损伤的活性氧物质,并且通常被视为与年龄相关的事件。在小鼠中增加抗氧化剂的表达对神经元中的活性氧物质具有保护作用,外源性补充抗氧化剂也是如此。锰超氧化物歧化酶需要锰来发挥其酶功能。抗氧化治疗被认为适用于与年龄相关的神经退行性疾病,一种新型的锰超氧化物歧化酶模拟物——avasopasem manganese被描述并提出作为减少引起神经退行性疾病的氧化应激的潜在治疗方法。本综述的目的是探讨氧化应激导致神经退行性损伤的证据以及抗氧化基因在抑制活性氧物质损伤中的作用。能否减少或逆转导致神经炎症和神经退行的氧化应激的神经环境?
Houldsworth A. Role of oxidative stress in neurodegenerative disorders: a review of reactive oxygen species and prevention by antioxidants. Brain Commun. 2024 Jan 2;6(1):fcad356.
全球范围内,神经系统疾病是第二大死亡原因和主要残疾原因。在英国,有85万人生活在难以治愈和难治性痴呆症中,每年的费用超过260亿英镑,预计到2040年将翻倍。因此,对神经退行性疾病的理解和新治疗方法的探索至关重要。
本综述旨在检查一些减少中枢神经系统(CNS)中活性氧物种(活性氧)和氧化应激(氧化应激)影响的细胞内抗氧化机制,并探讨这些机制的缺陷作为神经退行性疾病(ND)的病因,特别关注阿尔茨海默病、帕金森病和运动神经元疾病。研究文献的多个来源被探索,并且信息被综合起来,以提供这个话题的最新概览。
疾病的发病机制在很大程度上是由活性氧作用导致的蛋白质理化性质改变所致。本综述的重点是描述抗氧化酶超氧化物歧化酶(SOD)在减少氧化应激中的作用。ND的特点是由于氧化应激造成的神经元损伤,论文中还讨论了一些炎症过程和表观遗传因素,它们降低了抗氧化功能。
尽管为这些疾病开处了几种药物,但有些药物无法穿过血脑屏障(BBB),它们在治疗ND中的有效性有限。
活性氧、氧化应激和神经退行性疾病
活性氧是一组具有奇数、未成对电子的原子团,导致氧化应激,并在ND的病理生理学中发挥重要作用。这可能因线粒体功能障碍或抗氧化基因表达降低而加剧。自由基(R•s)的几个例子包括超氧阴离子、氧、羟基、烷氧基和过氧自由基,以及一氧化氮和二氧化氮(图1)。还有非自由基活性氧,如过氧化氢、次氯酸和几种氮化合物。氧化应激生物标志物检测工具可用于调查ND,例如免疫荧光。使用硝基酪氨酸、4-羟壬烯醛和8-羟基鸟嘌呤在海马切片中是检测方法的一个例子。其他可能作为诊断工具有用的生物标志物可能是蛋白质羰基。
衰老的自由基理论是一个确立已久的理论,它提出由活性氧物种(活性氧)引起的氧化损伤是衰老的主要原因。与神经退行性疾病(ND)相关的活性氧效应在阿尔茨海默病、帕金森病、亨廷顿病、肌萎缩侧索硬化症、多发性硬化症和弗里德里希共济失调等疾病中已被充分证实。事实上,活性氧已被证明能引起基因甲基化,改变遗传表观遗传景观。许多慢性疾病是由环境暴露触发的,例如活性氧,它们可以在表观基因组中引起异常变化,并可在慢性疾病中重塑DNA甲基化。
与大脑中的氧化应激(氧化应激)强烈相关的APOE4基因型已被认定为阿尔茨海默病表型和发病机制的易感基因。ApoE4是ApoE家族(ApoE1-4)中在应对氧化应激方面功能最弱的抗氧化剂,其参与SOD2抗氧化功能将在后文讨论。
尽管存在外源性和内源性来源,但非常活跃的内源性生物活性氧的主要来源是线粒体,在线粒体电子传递过程中,超氧阴离子自由基、过氧化氢和羟基自由基在氧气还原为过氧化氢的过程中产生(图2)。内源性活性氧还通过免疫细胞激活、炎症、精神压力、过度运动、缺血、感染、癌症和衰老产生,而外源性来源包括污染、酒精、烟草、烟雾、重金属、过渡金属、工业溶剂、农药、辐射和某些药物。
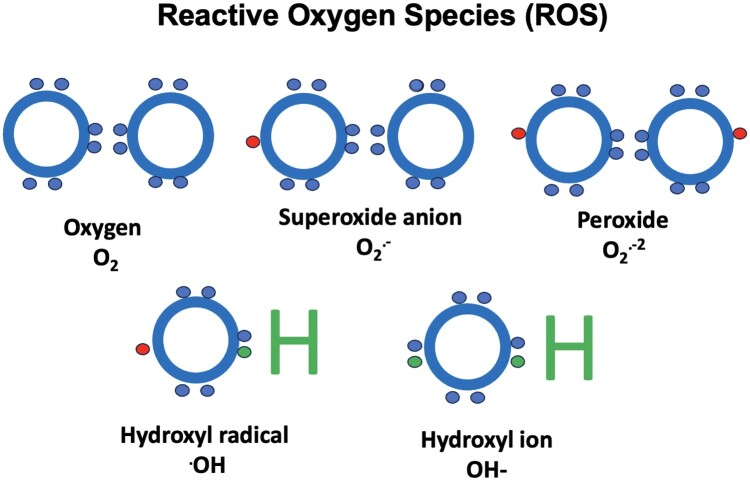
图1 作为正常氧气代谢副产物的活性氧(活性氧),包括过氧化物、超氧阴离子、羟基自由基和羟基离子。所有这些物种都有一个未配对的电子(除了羟基离子),这使得它们非常不稳定,导致氧化应激,可以造成细胞结构和DNA的损伤。然而,一些低水平的活性氧可以在细胞内信号传导中发挥作用。

图2.内源性和外源性来源活性氧的产生。活性氧引起氧化应激,可以造成DNA和组织损伤,导致疾病过程。抗氧化基因,如SOD2,可以减少氧化应激,使细胞修复发生,并减少由氧化应激引起的疾病的发病机理。氧化应激可以损伤中枢神经系统和脑细胞,导致与NDG相关的疾病,如阿尔茨海默病、帕金森病和运动神经元疾病-这些疾病分别在大脑图中由大脑的蓝色和紫色区域表示。据推测,减少氧化应激可以抑制NDG的发病机理。
酶促反应包括由吞噬作用和线粒体呼吸链产生的那些,它们与非酶促过程不同,例如有氧呼吸中的氧化磷酸化。然而,活性氧的其他次要来源包括过氧化物酶体和内质网,以及各种分解代谢途径。这些潜在的危险活性氧化中间体作为正常代谢的副产品,在几种疾病包括神经退行性疾病(NDG)的发病机制中可以发挥重要作用。神经和血管功能障碍也可以通过细胞质中游离NADH/NAD+比率增加导致的假性缺氧来介导,与真性缺氧类似。线粒体中活性氧的过量产生可以导致线粒体脂质、蛋白质和DNA的氧化。此外,众所周知,氧化应激还与蛋白质错误折叠有关,如在Creutzfeldt–Jakob病中所观察到的。然而,活性氧的适度增加可以激活积极的细胞反应,称为毒物兴奋效应,并且可以对抗正常衰老过程,与促进炎症、癌症和细胞死亡的信号通路的高度激活相比,后者在加速衰老表型中观察到。这是一个例子,说明低剂量的化学剂或环境因素暴露是有益的,但在高剂量下则有害。
毒物兴奋效应是一个低剂量的化学剂或环境因素暴露有益但高剂量有害的例子,其中适度增加的活性氧可以激活积极的细胞反应。因此,尽管活性氧失衡可能导致疾病,这些高度活性的物种也是具有多种生理功能的必需分子,有时在许多组织中充当第二信使。因此,当活性氧以低水平存在时,它们在神经系统中具有重要的信号分子作用,但如果它们的水平升高到破坏其稳态的程度,它们也在炎症性疾病的进展中发挥作用。当多形核白细胞在免疫应答过程中在炎症部位产生活性氧时,可能会导致组织损伤和内皮功能障碍。有证据表明活性氧可能影响神经分化的多个方面,其中亚致死水平的自由基和过氧化氢影响了细胞内信号传导途径。这些机制调节了几种不同的神经元和非神经元细胞中的基因表达、细胞分化和生长。一项研究表明,正常的活性氧产生调节了神经元成熟在生化、生理和形态上的过程,其中一些过程特别是由超氧阴离子介导的。微胶质细胞和小胶质细胞可以被这些活性物质激活。
还必须注意的是,线粒体活性氧的产生是先天免疫系统中对细菌、病毒和真菌感染的有价值的杀菌武器。这些高度活跃的活性氧在靶向上是不加选择的,因此除了病原体外,还会对局部组织细胞造成损伤。这种在感染期间触发免疫应答而产生的活性氧可以损伤健康细胞并诱发慢性炎症,包括神经炎症(NI)和退行性变。
由于氧化应激而激活的炎性小体与积聚在细胞质中的多蛋白复合物相关,并导致炎性过程和功能障碍性细胞清除。
大脑某些区域含有高铁量的另一个氧化应激来源可以刺激诸如超氧阴离子和过氧化氢的自由基反应。铁复合物的与年龄相关的积累可能是诊断ND的潜在生物标志物工具。大脑中铁的积累需要严格调节,以防止它产生可以通过表观遗传机制影响DNA表达的活性氧。
抗氧化剂
一般来说,抗氧化剂是天然的多羟基化酚类化合物,分子量低。一些细胞酶在细胞室内表达,具有强大的抗氧化特性,可以消除自由基。许多蔬菜和水果也含有具有抗氧化特性的饮食多酚,包括黄酮类、酚酸、鞣质、木脂素、芪类、儿茶素和类胡萝卜素。因此,很明显抗氧化剂可以防止分子的细胞内氧化。在这个过程中,电子或氢从物质中移除,因此,它们可以通过直接与自由基反应来减少对细胞的氧化损伤。认为这些抗氧化物质的芳香环上的羟基位置和数量可能在抗氧化活性中发挥重要作用。因此,抗氧化剂是可以减少活性氧引起的氧化损伤的自由基清除剂。通过抗氧化剂中和活性氧可以是内源性或外源性的。
超氧化物歧化酶
催化超氧阴离子歧化的普遍存在的SOD酶家族可以根据其功能与铜、锰(Mn)、锌和铁相关联,其中铜/锌相关的SOD1在细胞质和细胞器中活跃。SOD3是一种胞外酶,SOD2在线粒体中活跃,具有几个结构差异(图3)。SOD2酶的基因位于第6号染色体上,与阿尔茨海默病有关的第6号染色体异常实际上与120多种其他疾病有关。SOD2具有金属酶抗氧化活性,依赖于过渡金属Mn,当新合成SOD2时,Mn被插入到复合物中。SOD2中和超氧自由基并将其转化为过氧化氢,控制线粒体中的二氧毒性,这是一个极端氧化负荷的细胞器(图4)。Mn是一个研究广泛的过渡金属,测量到患有阿尔茨海默病的患者Mn水平显著降低,并与轻度认知障碍有关。还有其他关键酶依赖于Mn,如谷氨酸合成酶、精氨酸酶和丙酮酸羧化酶。
SOD2是一种抗氧化酶,可以催化超氧阴离子的歧化反应,从而减少氧化应激。在小鼠中,SOD2的过表达可以减少海马区的超氧化物并防止记忆缺陷。然而,人类随着年龄的增长,抗氧化基因的表达会降低,这在38岁以上女性的卵泡中SOD1、SOD2和过氧化氢酶的mRNA表达降低中可以看出,这表明了对活性氧的防御能力随着年龄的增长而减弱。
SOD2的表达也受到基因变异的影响。例如,SOD2基因的一些多态性,如V16A多态性(rs4880),与SOD2野生型相比表达降低,被认为是由氧化应激引发的各种疾病的易感基因。然而,关于SOD2基因型与阿尔茨海默病的关联存在一些不一致性,一些研究者认为SOD2(rs4880)在阿尔茨海默病中并没有像SOD1在帕金森病中那样具有决定性作用。
褪黑素是一种可以改善氧化应激并增加SOD2表达的抗氧化剂。当给予辐射组织时,褪黑素可以提高正常组织的SOD2活性,但对癌症组织无效。此外,褪黑素补充可以对抗氧化活性产生积极影响。
镉被发现可以抑制酶的抗氧化特性,如SOD2和过氧化氢酶。它还会引起突变和染色体缺失,同时增强活性氧的产生。人体中外源性镉的主要来源是吸烟,据报道30%的吸烟者会发展为血管性痴呆,40%的人会发展为阿尔茨海默病。
目前,临床上使用SOD2的例子还很少。考虑到SOD2疗法的概念,通过口服给药的外源性SOD2补充可能会因为对外源蛋白产生抗药抗体或蛋白质在被吸收前被消化而变得复杂,因此正在研究新的给药方法。肌肉内注射SOD2是一种可能性。除了补充SOD2外,还提出了通过相关转录因子增强沉默基因的去甲基化或增强转录。一种叫做orgotein的牛源SOD可以通过肌肉内注射给药,但可能含有一些引起过敏反应的污染物。最近的药物进展产生了avasopasem Mn(GC4419),一种SOD模拟物,可以选择性地将超氧阴离子还原为过氧化氢分子。该药物由Galeria开发。已经进行了avasopasem Mn作为放射治疗后口腔粘膜炎和食道炎治疗的临床试验,该药物似乎具有良好的耐受性,并发现可以保护正常组织;在临床试验中,该药物是通过输液给药的。
年龄相关的和进行性的神经退行性疾病涉及到共济失调和痴呆,影响到受这些疾病影响的人的寿命和生活质量。神经元和胶质细胞更容易受到氧化应激的影响,并且具有更高的代谢需求。这与身体其他细胞相比再生率较低,抗氧化能力不足,使得中枢神经系统容易受到氧化损伤。因此,抗氧化状态差或氧化剂和抗氧化剂平衡失衡与神经退行性疾病的发病机制有关。大脑组织代谢活跃,依赖氧化磷酸化作为其能源,并且含有高水平的脂质,这也消耗了大量的氧气。因此,氧化应激可以影响神经元的功能和存活。广泛的神经退行性疾病与这种抗氧化剂失调有关。
线粒体蛋白质稳态基因调节蛋白质的伴侣、折叠和维护功能,随着年龄的增长,这些基因被下调,无法修复的蛋白质损伤积累起来。急性的活性氧积累和抗氧化防御机制之间的失衡导致氧化应激,如前所述,这被认为在阿尔茨海默病的发病机制中起着主要作用,其中失衡促进线粒体基因的表达,涉及代谢和活性氧的产生导致线粒体DNA中的基因突变。
神经炎症是一系列反应和免疫事件的级联,导致神经退行性疾病,一些证据表明氧化应激和神经炎症都有助于神经退行性疾病的病理生理学、发病和进展。氧化应激可以导致神经失调和炎症。正如下一节所讨论的,肥大细胞(MC)激活涉及细胞质颗粒的脱颗粒,其中包含一些有助于氧化应激的炎性介质(图1)。
神经退行性疾病可以特征化为特定神经元群体由于微管相关蛋白tau的异常构象而逐渐功能障碍,其中tau是一种稳定神经元内部骨架的蛋白质。Tau病变与许多神经退行性疾病有关,其中最常见的是阿尔茨海默病。氧化应激是神经退行性tau病变病理生理学的一个重要因素。Tau过度磷酸化与不溶性聚集体的形成、神经纤维缠结、突触功能障碍和神经元死亡有关,而这可以由氧化应激引起,因为它可以诱导Tau过度磷酸化。
免疫学中的神经退行性疾病 (NDG)
中枢神经系统(CNS)是一个免疫特权部位,外周免疫细胞通常不能穿过血脑屏障(BBB),但与其他许多造血免疫细胞不同,肥大细胞(MCs)存在于人脑中,并在介导感觉或神经内分泌区域的几个结构中找到,同时与神经免疫系统相互作用。
在病原体入侵期间,MCs是第一反应者,而活化的小胶质细胞和某些细胞因子的存在被报道为神经免疫(NI)中的关键角色。 MCs作为催化剂,启动并放大免疫和神经反应,招募其他炎症因子。它们还存在于血液-脑脊液屏障和脑膜的硬脑膜层。在大脑中,它们是BBB脑侧的效应器和传感器,在那里它们与小胶质细胞和星形胶质细胞相互作用。NI是一种导致NDG进展、功能障碍和神经元丢失的机制。
应激相关的NDG期间会释放促肾上腺皮质激素释放激素,由MCs释放,有助于下丘脑-垂体-肾上腺轴。因此,MCs在免疫系统和神经元之间进行交互。释放这种激素也是对应激的反应,并诱导MCs脱颗粒,这可能会破坏BBB并导致神经退行性疾病(ND)。
大脑中的这些炎症机制可以驱动淀粉样蛋白和β-淀粉样蛋白斑块的形成,这增强了痴呆和阿尔茨海默病的发病;tau形成突触毒素聚集体,发展成神经纤维缠结。作为CNS中的第一反应者,MCs脱颗粒形成趋化通路,为胶质细胞向病理刺激物移动。
小胶质细胞是CNS的先天免疫细胞,在NI中扮演着至关重要的角色。58-60 小胶质细胞是神经死亡细胞的清道夫,被认为参与大脑防御。它们是神经免疫系统的重要组成部分,大量参与清除β-淀粉样蛋白和发展NI。这一重要作用在阿尔茨海默病的发病机制中发挥作用。除了导致导致阿尔茨海默病发病机制的神经损伤外,还有研究表明小胶质细胞可能形成围绕淀粉样沉积物的保护屏障,其中淀粉样纤维被压缩,小胶质细胞充当管家吞噬细胞,通过促进Aβ清除来维持稳态。小胶质细胞也可能通过过量生成超氧阴离子而被激活。 然而,组织的抗氧化状态必须足够以应对这种增加的氧化应激。
尽管一些研究人员观察到小胶质细胞吞噬斑块并减少病理,但也有人认为它们可能负责传播病理,其中小胶质细胞的激活可以有助于NI,释放包括活性氧在内的炎症介质。已经识别出两种不同的小胶质细胞形式:稳态和疾病相关的小胶质细胞表型。 一些涉及调节小胶质细胞吞噬作用的新疗法已被提出。
帕金森病
在帕金森病的情况下,多巴胺能神经元的退化涉及氧化应激(氧化应激)。当生物过程打乱了神经元氧化还原电位的生理维持时,这可能导致细胞死亡。氧化和硝化损伤已在帕金森病的黑质中被确认。黑质中的多巴胺能神经元在帕金森病中减少。多巴胺是一种不稳定的分子,在大脑这个区域引起活性氧和多巴胺醌的产生,通过自动氧化引起氧化应激。这种氧化应激和线粒体功能障碍是帕金森病中NDG的核心要素。衰老通过减少抗氧化基因如SOD2、过氧化氢酶和谷胱甘肽过氧化物酶的活动和表达,降低了对抗氧化应激的抗氧化机制的能力。
多巴胺本身经历自动氧化,是一种不稳定的分子,形成醌和自由基(R•s)。 SOD2酶活性被多巴胺醌修饰,并对帕金森病有影响。当表达多巴胺的黑质脑细胞丢失时,这会导致帕金森病。 中脑后部黑质致密部的多巴胺能神经元负责多巴胺的产生。基底神经节神经元控制运动,当它们死亡或受损时,会减少到身体的电信号。SOD2 V16A多态性(rs4880)与中国人群中帕金森病易感性的增加有关。这个V16A多态性比野生型基因的Mn SOD表达更低,并且已被证明与其他疾病状态相关,例如糖尿病肾病。给予功能性SOD2可能对帕金森病患者有益,减少与这些患者NG相关的氧化应激免疫病理学。
运动神经元疾病
人类有两种公认的运动神经元疾病形式,一种是家族性的,另一种是后天性的。家族性疾病与几个基因有关,但主要是SOD1突变。SOD1是一种细胞外的抗氧化剂,将R•氧物种还原为过氧化氢。家族性疾病出现在约12-20%的遗传基因缺陷中,1-2%是SOD1蛋白的自发突变,并且是散发性的。
最近的治疗方法是用一种针对该蛋白质的单克隆抗体tofersen(BIIB067)中和功能失调的肌萎缩侧索硬化症基因产品SOD1。使用m-RNA阻止异常SOD1蛋白功能的试验取得了显著改善疾病的进展和严重程度。2021年报告了针对SOD1突变患者的第三阶段试验(VALOR试验)的临床效益。
如前所述,大脑组织中铁的积累产生活性氧,需要严格调节,这可能是由功能失调的SOD1酶产生的。与SOD2不同,生物金属酶SOD1依赖于铁或铜才能发挥功能。
这些酶的抗氧化功能对于减少活性氧引起的氧化应激至关重要,从而抑制引起神经元损伤的异常蛋白的产生。在SOD2缺乏的小鼠中,存在与年龄相关的运动神经元疾病相关的信号改变。 SOD2敲除小鼠具有早期致死性和NDG,但Cre-Lox技术使得可以在神经元中减少SOD2。
因此,是否有可能纠正非功能性或异常形式的SOD1和SOD2的表达,以减少活性氧及其相关病理?或者,是否可以用外源性功能性SOD1进一步增强目前对家族性肌萎缩侧索硬化症患者的治疗?
阿尔茨海默病
阿尔茨海默病的发病机制被认为是 活性氧 产生过多的结果,除了 Aβ 的淀粉样斑块聚集外,还导致神经原纤维缠结作为 tau 聚集体。这也可能导致更高的代谢需求和/或线粒体功能障碍。
如前所述,阿尔茨海默病的标志包括 tau 蛋白和淀粉样蛋白沉积,在一项研究中,SOD1 表达的 tau 蛋白增加,在动物研究中,发现野生型 SOD1 基因表达对 NDG 具有保护作用。众所周知,家族性肌萎缩侧索硬化症是由 SOD1 (Cu/Zn) 突变引起的。事实上,中枢神经系统的几种慢性神经退行性病变具有一些共同特征,例如炎症、氧化应激、突触功能障碍、蛋白质错误折叠和有缺陷的自噬。
如前所述,大脑会产生大量的 活性氧,因为它是一个高度代谢活跃的器官,通常由精心设计的抗氧化剂网络保护,以维持微妙的平衡;因此,氧化应激被认为有助于阿尔茨海默病的发病机制。前面也提到过,一些阿尔茨海默病患者的Mn状态降低,SOD2的抗氧化功能依赖于这种过渡金属,因此,可能是支持性证据,表明 SOD2 功能可能是阿尔茨海默病发病机制的重要因素。
表观遗传景观和衰老对抗氧化机制的影响
表观遗传变化通过DNA的甲基化导致基因被不恰当地沉默,从而改变了遗传物质的可及性和蛋白质的表达。组蛋白也可以通过非编码RNA进行修饰,也称为垃圾RNA,并且可以通过表观遗传进行改变。在阿尔茨海默病精神病患者的死后脑样本中,已经注意到甲基组学变化。随着年龄的增长,可以观察到高甲基化,并且可以通过生化测试将其识别为表观遗传时钟的生物标志物,老年科学试图通过这些标志物来测量患者的表型年龄。由于与年龄相关的表观遗传景观的这些变化,其中一些是DNA反应性氧化损伤的结果,氧化应激的积累会导致认知衰老和NDG。SOD2 已被证明受到这种机制的影响和修饰,尤其是因为糖尿病。SOD2 启动子内 CpG 核苷酸的高甲基化与 SOD2 表达降低有关。
正如之前所提到的,APOE4是阿尔茨海默病的一个易感基因,具有抗氧化活性。在证明抗氧化功能作用的实验中,APOE4靶向替换小鼠表现出更高的抗氧化活性以抵抗氧化应激。如果其他抗氧化剂(如SOD2)的表达也被沉默或因年龄而减少,表观遗传改变可以解释与阿尔茨海默病发病机制相关的增加的氧化应激。此外,尽管关于SOD2基因rs4880-T等位基因与阿尔茨海默病的直接关联有一些讨论,但轻度认知障碍和阿尔茨海默病患者在APOEe4等位基因和SOD2多态性组合的人群中更为普遍。
结论
从研究中可以清楚地看出,活性氧生成的失衡可能是神经组织中氧化应激的原因,但有效的抗氧化功能也可以减少由活性氧引起的氧化损伤(表1)。抗氧化机制显然有可能通过消除活性氧来减少氧化应激;然而,抗氧化基因的表达可能受到年龄、表观遗传事件以及DNA损伤机制的影响,从而降低其减少活性氧的能力。此外,睡眠剥夺可能会降低褪黑激素的作用,褪黑激素是SOD2的激活因子。
在提出所有这些证据之后,必须考虑改变中枢神经系统微环境的问题,以防止发生不可逆的损害,尤其是与活性氧和氧化应激致病水平相关的损害,以及这些损害是否可以逆转。希望这篇综述能有助于对氧化应激诱导的神经退行性疾病的临床前研究增进了解,从而提高临床转化治疗水平。
技术进步可以改进应对氧化应激诱导疾病的策略,而最近的纳米技术时代为药物穿越血脑屏障提供了新的机会,这主要涉及打开紧密连接和抑制外排泵。除了抗氧化剂类似物外,增加抗氧化酶活性的方法也可以减少神经组织的氧化损伤,如在阿尔茨海默病、帕金森病和运动神经元疾病中观察到的。SOD2表达的增加和具有相似功能的类似物已被证明可以减少活性氧并防止氧化应激在其它原因引起的氧化应激的临床试验中,如放疗引起的组织氧化损伤。模拟SOD2活性的新类似物药物在减少放疗引起的氧化应激造成的损伤方面取得了成功,也可能能够减少神经退行性疾病的进展。希望通过增强抗氧化功能来测试这一假设的临床试验能够对一些ND有效。
神经退行性疾病的早期诊断正在改善,尤其是在发现易感基因以及在阿尔茨海默病、帕金森病和运动神经元疾病患者中观察到的不同基因表达组合之后。希望新时代的早期诊断和减少氧化应激可以减少一些神经退行性疾病在疾病早期阶段的发病机理。为增进对这个话题的理解,提出了一些建议,包括建立阿尔茨海默病患者的抗氧化状态、基因分型相关基因多态性、测量患者抗氧化状态以及检测患者锰、铜和锌状态以及氧化应激标志物。对一些神经退行性疾病患者进行SOD2类似物的临床试验将是评估其在患者早期诊断阶段疗效的一个有趣步骤。
https://wap.sciencenet.cn/blog-41174-1420227.html
上一篇:高效产氢气医用材料研究【西交+华师】
下一篇:改写教科书的新现象:心率相关脑电震荡