博文
Angew. Chem. Int. Ed.封面文章——Metal-Free Catalytic Hydrogenation(October 22, 2007)
|



|
Metal-Free Catalytic Hydrogenation
 |
| Preston A. Chase, Gregory C. Welch, Titel Jurca, Douglas W. Stephan, Prof. Dr. * |
| Department of Chemistry and Biochemistry, University of Windsor, Windsor, Ontario, N9B 3P4, Canada, Fax: (+1) 519-973-7098 |
| email: Douglas W. Stephan (stephan@uwindsor.ca) |
Supporting information for this article is available on the WWW under http://www.wiley-vch.de/contents/jc_2002/2007/z702908_s.pdf or from the author.
| Article Text |
Hydrogenation is the addition of hydrogen to unsaturated organic compounds. Such reactions are used for the production of a myriad of chemical products worldwide, from large-scale operations including the upgrading of crude oil and the production of bulk commodity materials to the synthesis of a variety of fine chemicals used in the food, agricultural, and pharmaceutical industries.[1] The process of hydrogen addition to unsaturated precursors is mediated by either homogeneous or heterogeneous transition-metal-based catalysts.[1] Blaser et al.[2] place the importance of this chemistry in context, stating that  hydrogen is the cleanest reducing agent and hydrogenation is arguably the most important catalytic method in synthetic organic chemistry both on the laboratory and the production scale
hydrogen is the cleanest reducing agent and hydrogenation is arguably the most important catalytic method in synthetic organic chemistry both on the laboratory and the production scale . Historically, hydrogenation began with Sabatier's 1897 discovery that traces of nickel could mediate the catalytic hydrogenation of olefins and culminated in a share of the 1912 Nobel Prize with Grignard. In the 1960s, the advent of organometallic chemistry gave rise to homogeneous transition-metal-based hydrogenation catalysts for a variety of substrates. The operation of these catalysts hinges on the key step of oxidative addition of hydrogen.[3] More recently, transition-metal systems that effect heterolytic cleavage of hydrogen at a metal center have been uncovered. In these cases, a metal hydride is formed with concurrent protonation of an amido ligand.[4], [5]
. Historically, hydrogenation began with Sabatier's 1897 discovery that traces of nickel could mediate the catalytic hydrogenation of olefins and culminated in a share of the 1912 Nobel Prize with Grignard. In the 1960s, the advent of organometallic chemistry gave rise to homogeneous transition-metal-based hydrogenation catalysts for a variety of substrates. The operation of these catalysts hinges on the key step of oxidative addition of hydrogen.[3] More recently, transition-metal systems that effect heterolytic cleavage of hydrogen at a metal center have been uncovered. In these cases, a metal hydride is formed with concurrent protonation of an amido ligand.[4], [5]
Non-transition-metal catalysts for hydrogenation reactions are all but unknown. KOtBu has been shown to act as a catalyst effecting the addition of H2 to benzophenone under forcing conditions of 200 癈 and greater than 100 bar H2.[6] Organocatalysts have been developed for hydrogenations of enones and imines; however, such systems do not employ H2 directly but rather a surrogate such as a Hantzsch ester as the stoichiometric source of hydrogen.[7]-[11] The development of nonmetal hydrogenation catalysts hinges on the discovery of systems that react cleanly with H2, but few are known. Power and co-workers reported the hydrogenation of Ge2-alkyne analogues to give a mixture of Ge2 and primary germane products.[12] Recently we have introduced the concept of frustrated Lewis pairs, bulky Lewis acids and bases which are sterically precluded from forming simple Lewis adducts.[13] Mixtures of frustrated phosphines and boranes can heterolytically cleave H2, forming phosphonium borates of the form [R3PH][BHR 3].[14] In very recent work, Bertrand and co-workers have demonstrated that selected carbenes exhibit transition-metal-like reactivity and cleave hydrogen or ammonia to effect a formal oxidative addition of the carbene C atom.[15] Last year, we reported the only nonmetal system known to reversibly activate and liberate H2. The phosphonium borate (2,4,6-Me3C6H2)2PH(C6F4)BH(C6F5)2 (1) is formed by reaction of the phosphine-borane species (2,4,6-Me3C6H2)2P(C6F4)B(C6F5)2 (3) with H2 while heating of the zwitterion 1 above 100 癈 liberates hydrogen and regenerates the phosphine-borane 3.[16] Herein, we demonstrate that this system and a related system provide the first metal-free hydrogenation catalysts that effect the addition of molecular H2 to imines, nitriles, and aziridines to produce primary and secondary amines in high yields under relatively mild reaction conditions.
3].[14] In very recent work, Bertrand and co-workers have demonstrated that selected carbenes exhibit transition-metal-like reactivity and cleave hydrogen or ammonia to effect a formal oxidative addition of the carbene C atom.[15] Last year, we reported the only nonmetal system known to reversibly activate and liberate H2. The phosphonium borate (2,4,6-Me3C6H2)2PH(C6F4)BH(C6F5)2 (1) is formed by reaction of the phosphine-borane species (2,4,6-Me3C6H2)2P(C6F4)B(C6F5)2 (3) with H2 while heating of the zwitterion 1 above 100 癈 liberates hydrogen and regenerates the phosphine-borane 3.[16] Herein, we demonstrate that this system and a related system provide the first metal-free hydrogenation catalysts that effect the addition of molecular H2 to imines, nitriles, and aziridines to produce primary and secondary amines in high yields under relatively mild reaction conditions.
The reduction of imines and nitriles is one of the best synthetic methods to generate secondary and primary amines, and has found tremendous importance in the pharmaceutical and fine chemicals industry.[17]-[20] The air- and moisture-stable phosphonium borates (R2PH)(C6F4)BH(C6F5)2 (R=2,4,6-Me3C6H2 (1)[16] and tBu (2)[13]) are active catalysts for the hydrogenation of C N multiple bonds with H2. For example, imines are reduced in toluene to the corresponding amines cleanly and in high yield at slightly elevated temperatures (80-140 癈) and H2 pressures (1-5 atm) in sealed glass bombs (Table 1, entries 1-6). The amine products are readily separated from residual catalyst by filtration through a plug of silica gel; no other side products are observed in the NMR spectra of the crude reaction mixtures. In the case of a sterically less demanding imine (Table 1, entry 6) no catalytic turnover was noted. Similarly, nitriles are not catalytically reduced, as these donors intervene in the catalytic cycle by binding strongly to the B center of the catalyst. Sequestering the N lone pair by coordination of Ph(H)C
N multiple bonds with H2. For example, imines are reduced in toluene to the corresponding amines cleanly and in high yield at slightly elevated temperatures (80-140 癈) and H2 pressures (1-5 atm) in sealed glass bombs (Table 1, entries 1-6). The amine products are readily separated from residual catalyst by filtration through a plug of silica gel; no other side products are observed in the NMR spectra of the crude reaction mixtures. In the case of a sterically less demanding imine (Table 1, entry 6) no catalytic turnover was noted. Similarly, nitriles are not catalytically reduced, as these donors intervene in the catalytic cycle by binding strongly to the B center of the catalyst. Sequestering the N lone pair by coordination of Ph(H)C NCH2Ph to B(C6F5)3 (Table 1, entry 7) allowed catalytic imine reduction to proceed. In a similar fashion, by employing the more active 2 as the catalyst, alkyl and aryl B(C6F5)3-bound nitriles are also successfully reduced and isolated as the corresponding primary amine-borane adducts (Table 1, entries 8-10).[21] In these cases, partial reduction of nitriles to the corresponding imines can not be intercepted or observed. It is noteworthy that the bis-borane adduct of adiponitrile is also fully reduced under similar conditions to give the bis-borane adduct of 1,6-diaminohexane (Table 1, entry 10). Attempts to reduce similar B(C6F5)3-isonitrile adducts were unsuccessful. However, catalytic reductive ring opening of an unactivated N-aryl aziridine functionality is achieved under similar conditions (Table 1, entry 11).
NCH2Ph to B(C6F5)3 (Table 1, entry 7) allowed catalytic imine reduction to proceed. In a similar fashion, by employing the more active 2 as the catalyst, alkyl and aryl B(C6F5)3-bound nitriles are also successfully reduced and isolated as the corresponding primary amine-borane adducts (Table 1, entries 8-10).[21] In these cases, partial reduction of nitriles to the corresponding imines can not be intercepted or observed. It is noteworthy that the bis-borane adduct of adiponitrile is also fully reduced under similar conditions to give the bis-borane adduct of 1,6-diaminohexane (Table 1, entry 10). Attempts to reduce similar B(C6F5)3-isonitrile adducts were unsuccessful. However, catalytic reductive ring opening of an unactivated N-aryl aziridine functionality is achieved under similar conditions (Table 1, entry 11).
| [a] Standard conditions: 5 mol % catalyst, 4 mL toluene, ca. 5 atm H2. [b] 1 atm H2. [c] Determined by 1H NMR spectroscopy. [d] 10 mol % catalyst. |
The relative rates of imine reductions shed some light on the mechanism of catalytic reduction. The sterically encumbered and electron-rich imine tBuNCPh(H) is reduced in one hour under 1 atm H2 pressure at 80 癈 in toluene with both catalysts 1 and 2 (Table 1, entries 1 and 2) whereas reduction of the electron-poor imine PhSO2NCPh(H) requires significantly longer reaction times (10.5-16 h), higher H2 pressure, and higher temperature (Table 1, entries 3 and 4). As these bulky imines and the corresponding amines do not form strong adducts with B(C6F5)3,[22] the relative rates of these reactions suggest that it is the basicity of the N center rather than the steric demand of the substituents that determines the rate of reduction. This observation indicates that imine reduction is initiated by proton transfer from P to N rather than by borohydride attack of the imine carbon center. To further address this question, stoichiometric reaction of the phosphonium borate Cy3P(C6F4)BH(C6F5)2[13] with the imine tBuNCPh(H) was performed. This resulted in no reaction even after heating to 120 癈 for 24 h, implying that catalysis is indeed initiated by imine protonation.
Additional mechanistic insight was gleaned from stoichiometric reactions of imines with 1 or 2. In an NMR tube, a 1:1 mixture of tBuNCPh(H) and 2 in CD2Cl2 immediately gives a yellow solution with a 31P NMR signal at  =22 ppm (compared to 35 ppm in 2), loss of the BH resonance in the 11B NMR spectrum, and a 19F NMR spectrum indicative of the formation of the neutral phosphine-borane 4 (Scheme 1). Species 4 forms a very weak adduct with the amine tBuNHCH2Ph. Similarly, the corresponding reaction of imine with 1 gives an orange solution which exhibits NMR parameters characteristic of the phosphine-borane 3 (Scheme 1).
=22 ppm (compared to 35 ppm in 2), loss of the BH resonance in the 11B NMR spectrum, and a 19F NMR spectrum indicative of the formation of the neutral phosphine-borane 4 (Scheme 1). Species 4 forms a very weak adduct with the amine tBuNHCH2Ph. Similarly, the corresponding reaction of imine with 1 gives an orange solution which exhibits NMR parameters characteristic of the phosphine-borane 3 (Scheme 1).
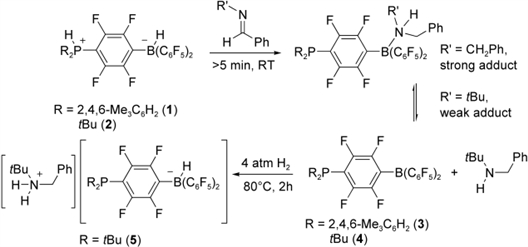
| Scheme 1. Stoichiometric reaction of imines with 1 or 2 with H2 in toluene. |
Given that we have independently established that 4 reacts with H2 to re-form 2, the stoichiometric reaction mixture of 2 and tBuNCPh(H) was treated with H2 pressure and subsequently heated to 80 癈 for 2 h, which discharged the yellow color and afforded the ion pair [tBuNH2CH2Ph][tBu2PC6F4BH(C6F5)2] (5, Scheme 1). This product is consistent with addition of H2 to 4; however, the greater basicity of the amine tBuNHCH2Ph over the phosphine center in 5 triggers proton transfer. Although it is possible that 5 is generated by the direct reaction of tBuNHCH2Ph and 4, the linked systems 3 and 4 react at a much greater rate with H2 (10 min) than the unlinked PR3/BR3 systems (12-24 h), suggesting that H2 is cleaved by 4 initially. Addition of excess imine to this mixture under a H2 atmosphere prompts smooth reduction to the corresponding amine, thus demonstrating that 5 acts as a living imine reduction catalyst. In a similar fashion, following complete reduction of PhSO2NCPh(H) with 5 mol % 1 (Table 1, entry 3), addition of another 20 equivalents of substrate prompted the continuation of imine reduction with no changes in reaction rate. A similar observation was made with the substrate 1,2,3-triphenylaziridine.
These data support a mechanism for hydrogenation of imines that is initiated by imine protonation[23] from the phosphonium borate zwitterion to give the iminium salt, which then undergoes nucleophilic attack by the borohydride anion, transferring hydride and affording the amine. Dissociation of the amine from the boron atom liberates the phosphine-borane, which then reacts with H2 to regenerate the phosphonium borate (Scheme 2). Similar proton initiation was found to be important for the B(C6F5)3-catalyzed ring opening of aziridines.[24]-[26]
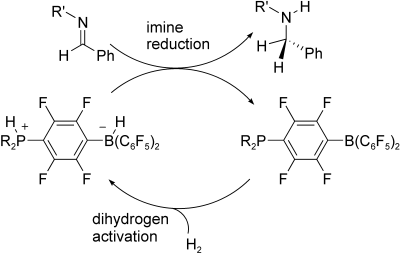
| Scheme 2. Catalytic cycle for reduction of imines. |
An important aspect of the mechanism involves suppression of catalyst inhibition by amine-borane adduct formation. In some cases, amine dissociation can be encouraged by the incorporation of sterically demanding substituents on the substrate that disfavor tight adduct formation. As well, elevated temperatures between 80-120 癈 promote amine dissociation and speed catalytic reduction. In contrast, while stoichiometric reductions are complete within 5 minutes at room temperature, catalysis is blocked by amine-borane adduct formation. Related steric effects have also been noted in the rates of imine hydrosilation catalyzed by B(C6F5)3.[22] In the case of the imine substrate PhCH2NCPh(H) (Table 1, entry 5) amine-borane adduct formation inhibits reduction, thereby allowing only stoichiometric reduction. The resulting amine (PhCH2)2NH coordinates strongly to the boron center of 4, as evidenced by NMR spectroscopy, even at elevated temperatures. However, sequestering the N-based lone pair by coordination to the Lewis acid B(C6F5)3[27]-[29] liberates the phosphine-boranes R2PC6F4B(C6F5)2 to activate H2, allowing the catalytic cycle to turnover and provide the amine-B(C6F5)3 species in reasonable yields. This protection strategy is based on our previous report demonstrating that B(C6F5)3 is a better Lewis acid than the species R2PC6F4B(C6F5)2 (R=2,4,6-Me3C6H2, tBu).[13] It is also important to note that coordination of the substrate to B(C6F5)3 prompts a mechanistic alteration. The reduction is initiated by hydride transfer from 1 or 2 to the imine carbon atom of the activated imine-borane adduct, in which B(C6F5)3 acts as a Lewis acid promoter, and is completed by proton transfer from the phosphonium center to the intermediate amido borate anion (Scheme 3).
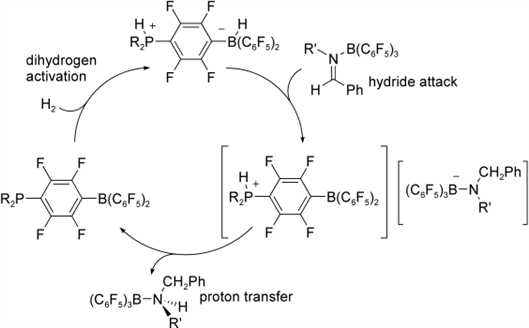
| Scheme 3. Catalytic cycle for reduction of B(C6F5)3-protected imines. |
While imines, nitriles, and aziridines are efficiently reduced in a catalytic manner, aldehydes react only in a stoichiometric fashion. For example, reaction of benzaldehyde with 1 or 2 occurs rapidly upon mixing at room temperature with reduction of the CO double bond, but the NMR spectra of the final product are consistent with the zwitterionic formulation R2PHC6F4B(C6F5)2OCH2Ph (R=2,4,6-Me3C6H2 (6) and tBu (7); Scheme 4). It is perhaps not surprising that proton transfer from the phosphonium center to the O atom does not occur, in contrast to imine reductions, as a result of the lower basicity of the O versus N atom. Nonetheless, efforts to achieve catalytic metal-free hydrogenation of carbonyl funationalities through the judicious design of related catalysts with both reduced Lewis acidity at boron and lowered basicity at phosphorus are in progress.

| Scheme 4. Stoichiometric reactions of benzaldehyde with 1 or 2 in CH2Cl2. |
This study demonstrates new and readily accessible metal-free catalyst systems for the reduction by H2 of imines, nitriles, and aziridines. These findings foreshadow a new battery of catalytic tools available for applications both in academic organic laboratories and in industry, allowing stoichiometric reductions mediated by NaBH4 or LiAlH4 to be performed in a catalytic fashion and greatly reducing subsequent waste production. Moreover, this approach could replace expensive precious-metal catalysts, thus offering the potential benefit of lower cost and diminished environmental impact from heavy-metal pollutants.
 |
 |
 |
All experiments were performed on double-manifold N2(H2)/vacuum lines or in a N2-filled MBraun 130-BG glove box. Toluene was dried by passage through alumina and molecular sieves in a commercially available solvent-purification system. H2 gas (Praxair) was dried by passage through a column of 50:50 activated molecular sieves and Dririte. NMR experiments were performed on a Bruker Avance-300 spectrometer at 300 K unless otherwise noted. 1H and 13C{1H} NMR spectra are referenced to SiMe4 using the residual proton peak of the given solvent. 31P, 11B, and 19F NMR spectra were referenced to 85 % H3PO4, BF3(OEt2), and CFCl3, respectively. Combustion analyses were performed in house employing a Perkin Elmer CHN analyzer. All imines, nitriles, and 1,2,3-triphenylaziridine were purchased from Aldrich and used without further purification.
Typical catalysis procedure: Each catalytic run was performed in duplicate, and the yields reported are the averages between the two runs. The reactions were monitored periodically by 1H NMR spectroscopy until complete. In all cases, the crude mixtures of the completed reactions were pure to the limits of NMR spectroscopy. a) In a glove box, a 100-mL glass bomb equipped with a small stir bar and a teflon screw tap was charged with imine (1 mmol), catalyst (0.05 mmol, 5 mol %), and dry toluene (4 mL). The reaction was transferred to the vacuum/H2 line and was degassed three times with a freeze-pump-thaw cycle. The reaction flask was cooled to -196 癈, 1 atm of H2 was introduced, and the flask then sealed and warmed to room temperature. The reaction was placed in a preheated oil bath and stirred at 500 rpm; at 120 癈, this gave an H2 pressure of about 5 atm. To take aliquots, the reaction was cooled rapidly in an ice bath, vented to release the H2 pressure, and taken into a glove box. b) In a glove box, the catalyst (0.05 mmol) was weighed into a 50-mL round-bottom Schlenk flask and slurried in toluene (2 mL). The reaction was attached to a vacuum/H2 line and subjected to three freeze-pump-thaw cycles. The slurry was then allowed to equilibrate at the desired temperature under an atmosphere of H2 with rapid stirring (500 rpm). A solution of substrate (1.0 mmol) in toluene (2 mL) was added by syringe. Aliquots were taken periodically by syringe. After reaction was complete, all volatiles were removed in vacuo, and the product was purified by trap-to-trap vacuum distillation or filtration through a small plug of silica (ca. 30 cm) with hexanes/ethyl acetate (3:1, 200 mL) eluent to remove residual catalyst. The B(C6F5)3-protected amines were not purified from residual catalyst.
| References |
|
|
|
| 1 | J. G. de Vries, C. J. Elsevier, The Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, 2007. |
| 2 | H. U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner, M. Studer, Adv. Synth. Catal. 2003, 345, 103. Links |
| 3 | R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, 2nd ed., Wiley, New York, 1994. |
| 4 | M. Yamakawa, H. Ito, R. Noyori, J. Am. Chem. Soc. 2000, 122, 1466. Links |
| 5 | K. Abdur-Rashid, A. J. Lough, R. H. Morris, Organometallics 2000, 19, 2655. Links |
| 6 | A. Berkessel, T. J. S. Schubert, T. N. Mueller, J. Am. Chem. Soc. 2002, 124, 8693. Links |
| 7 | P. I. Dalko, L. Moisan, Angew. Chem. 2004, 116, 5248-5286; Links Angew. Chem. Int. Ed. 2004, 43, 5138. Links |
| 8 | H. Adolfsson, Angew. Chem. 2005, 117, 3404; Links Angew. Chem. Int. Ed. 2005, 44, 3340. Links |
| 9 | M. Rueping, A. P. Antonchick, T. Theissmann, Angew. Chem. 2006, 118, 6903-6907; Links Angew. Chem. Int. Ed. 2006, 45, 3683. Links |
| 10 | J. B. Tuttle, S. G. Ouellet, D. W. C. MacMillan, J. Am. Chem. Soc. 2006, 128, 12662. Links |
| 11 | J. W. Yang, M. T. Hechavarria Fonseca, B. List, Angew. Chem. 2004, 116, 6829; Links Angew. Chem. Int. Ed. 2004, 43, 6660. Links |
| 12 | G. H. Spikes, J. C. Fettinger, P. P. Power, J. Am. Chem. Soc. 2005, 127, 12232. Links |
| 13 | G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. Wei, D. W. Stephan, Dalton Trans. 2007, 3407. Links |
| 14 | G. C. Welch, D. W. Stephan, J. Am. Chem. Soc. 2007, 129, 1880. Links |
| 15 | G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller, G. Bertrand, Science 2007, 316, 439. Links |
| 16 | G. C. Welch, R. R. S. Juan, J. D. Masuda, D. W. Stephan, Science 2006, 314, 1124. Links |
| 17 | M. L. Clarke, G. J. Roff, The Handbook of Homogeneous Hydrogenation (Eds.: J. G. de Vries, C. J. Elsevier), Wiley-VCH, Weinheim, 2007, p. 413. |
| 18 | F. Spindler, H.-U. Blaser, The Handbook of Homogeneous Hydrogenation (Eds.: J. G. de Vries, C. J. Elsevier), Wiley-VCH, Weinheim, 2007, p. 1193. |
| 19 | W. Tang, X. Zhang, Chem. Rev. 2003, 103, 3029. Links |
| 20 | S. Kobayashi, H. Ishitani, Chem. Rev. 1999, 99, 1069. Links |
| 21 | A. J. Mountford, S. J. Lancaster, S. J. Coles, P. N. Horton, D. L. Hughes, M. B. Hursthouse, M. E. Light, Inorg. Chem. 2005, 44, 5921. Links |
| 22 | J. M. Blackwell, E. R. Sonmor, T. Scoccitti, W. E. Piers, Org. Lett. 2000, 2, 3921. Links |
| 23 | J. B. Aberg, J. S. M. Samec, J. E. Bäckvall, Chem. Commun. 2006, 2771. Links |
| 24 | A. Caiazzo, S. Dalili, A. K. Yudin, Synlett 2003, 14, 2198. Links |
| 25 | I. D. G. Watson, L. Yu, A. K. Yudin, Acc. Chem. Res. 2006, 39, 194. Links |
| 26 | I. D. G. Watson, A. K. Yudin, J. Org. Chem. 2003, 68, 5160. Links |
| 27 | Imine and amine adducts of B(C6F5)3 have been previously reported.[22, 28, 29] |
| 28 | J. M. Blackwell, W. E. Piers, M. Parvez, R. McDonald, Organometallics 2002, 21, 1400. Links |
| 29 | W. E. Piers, Adv. Organomet. Chem. 2004, 52, 1. Links |
pdf全文下载
https://wap.sciencenet.cn/blog-3913-9115.html
上一篇:2007诺贝尔化学奖获得者Ertl早期在《德国应用化学》上有关界面化学综述——气固多相催化表征
下一篇:Chemists Strike Gold With New Gold Catalysts【旧闻回顾】