博文
Membrane Protein Simulation with Desmond
||
Desmond supports algorithms typically used to perform fast and accurate molecular dynamics. Long-range electrostatic energy and forces can be calculated using particle-mesh-based Ewald techniques.Constraints can be enforced using the M-SHAKE algorithm. These approaches can be used in combination with time-scale splitting (RESPA-based) integration schemes. Here are the generally steps for membrane protein simulation in Desmond.

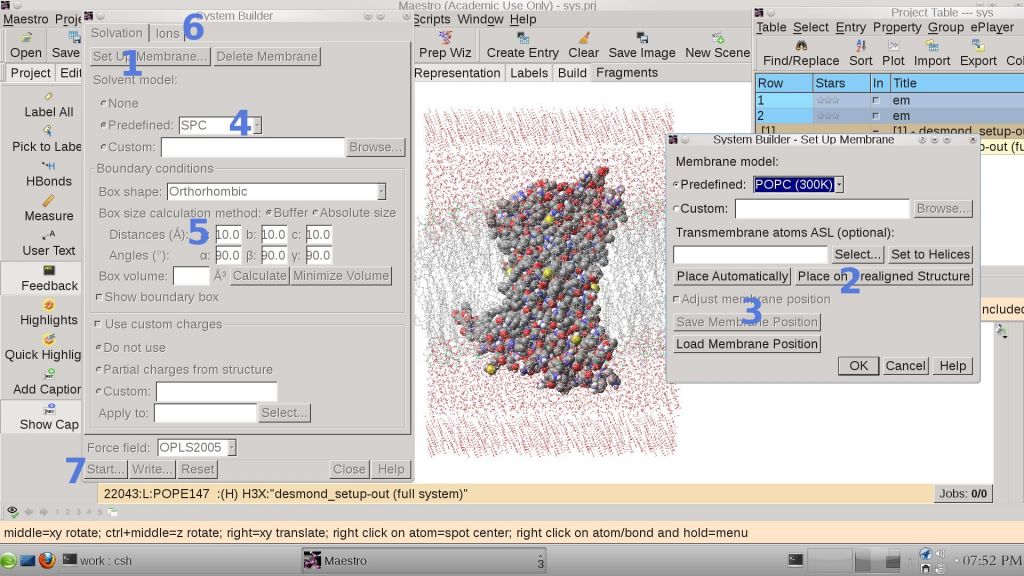
1. System Building
This can be done very easily with Maestro GUI from Schrodinger.
menu-->application-->desmond-->system builder
this will need 7 steps to be finished as shown in the snapshot. The membrane in Desmond are all pre-equilibrated which is quite important for initial membrane system. step 2 in the system building step is also quite important, for we can place the membrane in pre-aligned structures so that our protein can be embedded in correct origination in membrane.
The default size for membrane system is 10X10X10 A^3. It is necessary to adjust this according to the according to our membrane protein size. Sometimes, membrane system may be broken if the system is too small.
step6 can add salt to the system, namely 0.15 M NaCl usually,to mimic physiology environment.
Output files:
desmond_setup-out.cms
2. Assign Force Field
We can only assign OPLS_FF in the initial step. However, most of us would like to use other force field such as CHARMM or AMBER. In the latest version of Desmond, the following FFs are available:
charmm22nocmap
charmm27
charmm32
charmm36_lipids
amber94
amber96
amber99
amber99SB
amber99SB-ILDN
amber03
oplsaa_impact_2001
oplsaa_impact_2005
oplsaa_ions_Jensen_2005
tip3p
spce
tip3p_charmm
tip4p
tip4pew
tip5p
FF can be assigned by command:charmm27
charmm32
charmm36_lipids
amber94
amber96
amber99
amber99SB
amber99SB-ILDN
amber03
oplsaa_impact_2001
oplsaa_impact_2005
oplsaa_ions_Jensen_2005
tip3p
spce
tip3p_charmm
tip4p
tip4pew
tip5p
$SCHRODINGER/run -FROM desmond viparr.py [options] input-file output-file
for example:
$SCHRODINGER/run -FROM desmond viparr.py -f charmm27 -f tip3p desmond_setup-out.cms charmm.cms
Viparr uses atomic numbers and bond structure to match residues to templates. Thus if there are non-standard atom or residue PDB names in the .cms file, there is no need to modify them to match the names used in the force field.
You may also use multiple force fields when parameters assigned to a particular residue by one force field override parameters assigned by another force field. This situation is similar to combining force fields; residues in the chemical system match templates in more than one of the specified force fields and all matching force fields are applied. However, in this case two or more force fields provide parameters for the same term; for example, two force fields provide parameters for the angle between atoms 1, 2, and 3 causing a conflict. The conflict is resolved by allowing the first force field (that matches the residue) to take precedence. The order is the order in which the force fields were
specified on the command line.
3. Add Constraints
While the System Builder automatically adds all the necessary constraints to the .cms file, Viparr does not. The build_constraints.py script is all but mandatory; otherwise Desmond cannot make use of the SHAKE algorithm to constrain bonds between heavy atoms and hydrogens. The build_constraints.py script should be run on your .cms file after every use of viparr. Viparr removes the constraints each time.
If you process a .mae or .cms file with Viparr you will have to add the constraints in a separate step by running the following command:
$SCHRODINGER/run -FROM desmond build_constraints.py input.cms constraints.cms
4. Relax System
It can be difficult to relax freshly built protein-membrane systems. In particular, penetration of the water between the protein than the lipids can be problematic and require very lengthy simulations to correct. A relaxation protocol that should reduce or eliminate such problems is available by running the script relax_membrane.py from the command line.
a. Run the script to prepare the necessary input files:
$SCHRODINGER/run relax_membrane.py -i constraints.cms -t temperature -j protein-membrane
b. Run the membrane relaxation protocol using the command:
$SCHRODINGER/utilities/multisim -JOBNAME protein-membrane -HOST myhost -mode umbrella -cpu cpus -i protein-membrane-in.cms -m protein-membrane.msj -o protein-membrane-out.cms
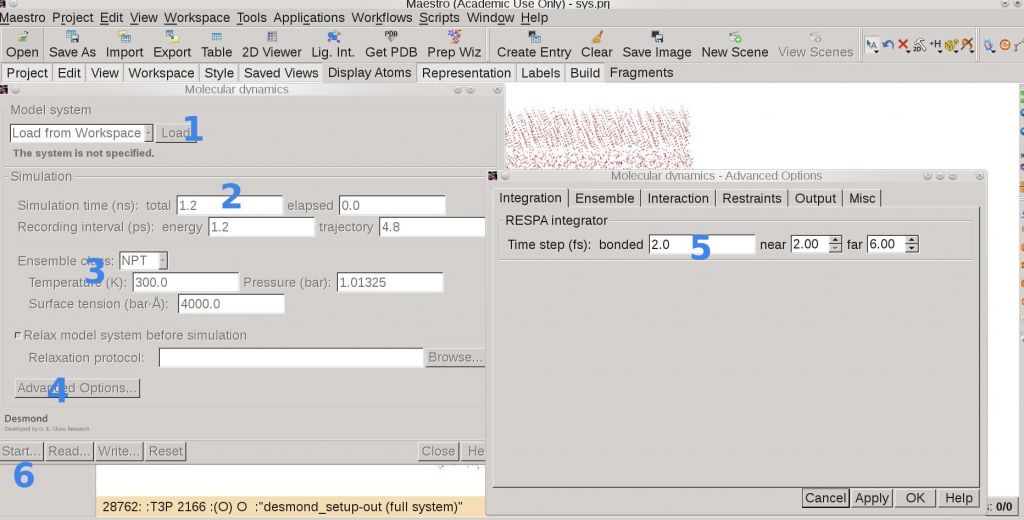
5. MD Production
Now we get the relaxed membrane system protein-membrane-out.cms at hand, and MD can be produced either by importing it to Maestro or by command line.
$SCHRODINGER/utilities/multisim -JOBNAME myMD -HOST my-16cpu -maxjob 0 -cpu "2 2 4" -m desmond_md_job.msj -c desmond_md_job.cfg -description "Molecular dynamics" -mode umbrella -i protein-membrane-out.cms -o desmond_md_job-out.cms -DISP append > log &
Tips:
We can setup host, CPU, tmp directory from /home/user/.schrodigner/schrodinger.hosts. Command line based jobs can be monitored from the tmp directory which we set in schrodinger.hosts file.
Reference
Desmond User Manual
Desmond Tutorial
Desmond User's Guide
download:
http://www.deshawresearch.com/downloads/download_desmond.cgi/
resource:
http://www.deshawresearch.com/resources_desmond.html
user group:
http://groups.google.com/group/desmond-md-users?hl=en
helps:
Desmond@DEShawResearch.com
https://wap.sciencenet.cn/blog-355217-458994.html
上一篇:humor at conference
下一篇:Desmond Configuration in Cluster