ВЉЮФ
ДњаЛбЇШЫ--NatureЃКЬЧдДњаЛЁЊЁАЬ№Ь№ЁБЕФжЌЗОЛсВњШШ
||
ДњаЛбЇШЫ
NatureЃКЬЧдДњаЛЁЊЁАЬ№Ь№ЁБЕФжЌЗОЛсВњШШ

зЋЮФ | ЩњУЏе§ ЙљЮФау еХД гкНЃ
БрМ | УЯУРбў
аЃЖд | гкНЃ
аЁБргаЛАЫЕ
ЬЧдЭЈГЃБЛШЯЮЊЪЧЯИАћжаФмСПДЂДцКЭРћгУЕФЪзбЁЁЃвШЕКЫиКЭЩіЩЯЯйЫиЖдЬЧдКЯГЩКЭНЕНтЕФЕїНкзїгУдкМЁШтКЭИЮдржавбгаЙуЗКЕФБЈЕРЃЌЕЋдкжЌЗОЯИАћжабаОПЕУНЯЩйЁЃБОбаОПЗЂЯжЪмЖљВшЗгАЗЕїНкЕФЬЧдДњаЛЖдUCP1БэДяЦ№живЊзїгУЁЃТ§адІТ-ЩіЩЯЯйЫиФмМЄЛюЕМжТБэДяUCP1ЕФжЌЗОЯИАћжаЬЧдЛ§РлдіМгЁЃPTGЃЈаЁБрзЂЃКProtein targeting to glycogenЃЌЕААзАаЯђЬЧдЃЌгЩPPP1RЃЈProtein Phosphatase 1 Regulatory Subunit , ЕААзСзЫсУИ 1 ЕїНкбЧЛљ)БрТыЁЃPTGдкЖдвШЕКЫиУєИаЕФзщжЏжаИпБэДяЁЃЭЈГЃЧщПіЯТЃЌPTGЪЧвдЕААзЕФаЮЪНЗЂЛгЙІФмЃЌЕБНтЪЭЦфгЩPPP1R3CБрТыЪБЃЌЛсНЋЦфзїЮЊбЧЛљНјааУшЪіЁЃЃЉШБЪЇЪЙУзЩЋжЌЗОЯИАћжаЕФЬЧдЫЎЦНКЭUCP1ЕФБэДяНЕЕЭВЂМѕШѕСЫКЎРфЛђІТ-ЩіЩЯЯйЫиФмЪмЬхДЬМЄЕМжТЗЪХжаЁЪѓЬхжиЯТНЕЕФГЬЖШЁЃЛњжЦЩЯЃЌЬЧдзЊЛЛЃЈturnoverЃЉФмЙЛВњЩњЛюадбѕЃЌМЄЛюp38 MAPKЧ§ЖЏUCP1ЕФБэДяЁЃвђДЫЃЌЬЧддкжЌЗОЯИАћжаНЋЬЧДњаЛгыВњШШСЊЯЕЦ№РДЃЌДгЖјЗЂЛгЙиМќЕФЕїНкзїгУЁЃ
ЭиеЙдФЖС
ЕААзАаЯђЬЧд
PTGдкПижЦЬЧдКЯГЩЗНУцЦ№живЊзїгУЁЃдкЬхФкЃЌPTGПЩвдЕїНкИЮдрЃЌМЁШтКЭжЌЗОЕШЖржжзщжЏжаЕФЬЧдЫЎЦНЃЌвдгеЕМЬЧдКЯГЩЁЃ1997ФъбаОПШЫдБЪзДЮдк3T3-L1ЯИАћжаЗЂЯжPTGЃЌЫќгыСзЫсЛЏУИМЄУИЁЂСзЫсЛЏУИ aКЭЬЧдКЯУИаЮГЩИДКЯЮяЃЌВЮгыЬЧдДњаЛЁЃ
дкВИШщЖЏЮяЯИАћжаЃЌЬЧдЪЧЦЯЬбЬЧЕФжївЊДЂДцаЮЪНЁЃПижЦЬЧдКЯГЩКЭНЕНтЕФЯоЫйУИЗжБ№ЪЧGS(Glycogen synthase, ЬЧдКЯУИ)гыGP (Glycogen phosphorylase, ЬЧдСзЫсЛЏУИ)ЁЃPTGПЩвджБНггыPP1ЁЂGSЁЂGPКЭСзЫсЛЏУИМЄУИНсКЯЃЌБэУїЦфПЩзїЮЊЬЧдКЯГЩЕФЗжзгжЇМмЁЃдкЬЧдПХСЃжаЃЌPTGгыPP1ЕзЮяУИаЮГЩИДКЯЮяЃЌзщзАВЮгыЬЧдДњаЛЕФЕААзжЪЃЌВЂЭЈЙ§PP1ЖдGSгыGPНјааШЅСзЫсЛЏЁЃGSдкШЅСзЫсЛЏКѓБЛМЄЛюЃЌGPдђЪЇЛюЁЃЭЈЙ§етжжЕїПиЃЌPTGПЩвдДйНјЬЧдКЯГЩВЂвжжЦЬЧдЗжНтЁЃвђДЫЃЌЯШЧАЕФбаОПЗЂЯждкИЮдрЛђжЌЗОЯИАћжаЃЌЙ§БэДяPTGПЩвдЯджјдіМгЯИАћжаЕФЬЧдЫЎЦНЁЃСэЭтЃЌдкЙЧїРМЁжаЃЌPTGвВОпгаВЛПЩКіЪгЕФзїгУЁЃЙЧїРМЁжаЕФIRF4ПЩвдЭЈЙ§PTGЕїПиМЁЬЧдКЌСПЁЂЕїНкМЁШтЕФЪЪгІадДњаЛЗДгІЃЌНјЖјгАЯьдЫЖЏФмСІЁЃ

ВЮПМЮФЯзЃК
[1]Zhu X et al.Adv Sci (Weinh). 2020 Aug1;7(19):2001502.
[2]Yang R et al.BiosciRep. 2015 May 1;35(3):e00207.
[3]Printen JA, Brady MJ, Saltiel AR.Science. 1997 Mar 7;275(5305):1475-8.
[4]Brady MJ et al.JBiol Chem. 1997 Aug 8;272(32):20198-204.
[5]Brady MJ, et al. Trends Endocrinol Metab.1999;10(10):408-413.
БГОАНщЩм
діЧПжЌЗОЯИАћВњШШБЛЙуЗКШЯЮЊЪЧвЛжждіМгФмСПЯћКФЁЂМѕЧсЬхжиЁЂИФЩЦвШЕКЫиЕжПЙЕФжЮСЦЗНЗЈЁЃWATЃЈWhite adiposetissueЃЌАзЩЋжЌЗОзщжЏЃЉзиЩЋЛЏЃЈWhite fat browningЃЌвВГЦЮЊЁАУзЩЋЛЏЁБЃЌbeigingЃЉгажњгкЕїНкДњаЛЮШЬЌЁЃWATЕФзиЩЋЛЏЬиеїЪЧГіЯжЁАУзЩЋЁБжЌЗОЯИАћЃЌЗЂЩњЯпСЃЬхИЛМЏЃЌВЂВњЩњЖрЗПжЌЕЮКЭUCP1 (Uncouple protein 1ЃЌНтХМСЊЕААз1)ЕФИпБэДяЁЃЫфШЛДцдкUCP1ЗЧвРРЕЕФВњШШЭООЖЃЌЕЋUCP1ВњШШЭООЖШдШЛЪЧзиЩЋКЭУзЩЋжЌЗОЯИАћВњШШЕФжївЊЛњжЦЁЃUcp1ЕФзЊТМЪмЖржжвђЫиЕїПиЃЌЦфжаАќРЈЭЈЙ§ІТ-ЩіЩЯЯйЫиФмЪмЬхЕФНЛИаЩёОМЄЛюЁЃбгГЄРфДЬМЄЛђЖрДЮзЂЩфІТ-ЩіЩЯЯйЫиФмМЄЖЏМСОљПЩЩЯЕїUcp1ЕФmRNAКЭЕААзБэДяЃЌВЂгеЕМiWATзиЩЋЛЏЁЃ
ЖјЬЧдзїЮЊФмСПДЂДцКЭРћгУЕФжївЊГЩЗжЃЌдкФмСПДњаЛЕФЙ§ГЬжаЭЌбљВЛПЩКіЪгЁЃвбгабаОПБЈЕРЃЌGS(Glycogen synthaseЃЌЬЧдКЯГЩУИ)зїЮЊЬЧдКЯГЩжаЕФЯоЫйУИЃЌПЩвдБЛЕААзМЄУИСзЫсЛЏЪЇЛюЃЌвВПЩвдгЩPP1 (Protein phosphatase 1ЃЌЕААзСзЫсУИ1)ШЅСзЫсЛЏМЄЛюЁЃЖјGP(Glycogen phosphataseЃЌЬЧдСзЫсЛЏУИ)ЪЧЬЧдЗжНтЕФЯоЫйУИЃЌЫќДпЛЏЬЧдЕФСзЫсНтзїгУЃЌЪЙЬЧдЗжзгДгЗЧЛЙдЖЫж№ИіЖЯПЊІС-1ЃЌ4-ЬЧмеМќвЦШЅЦЯЬбЬЧЛљЃЌДгЬЧджаЪЭЗХГі1-СзЫсЦЯЬбЬЧЁЃGPКЭ GS ЛюадЕФЯрЛЅБфЛЏВПЗжЪЧгЩЕААзСзЫсУИ-1ЕФЬЧдАаЯђЕААзЃЈР§ШчБОЮФбаОПЕФPTGЃЉНщЕМЕФЃЌетаЉЕААзе§ЯђЕїНк GS ЕФЛюадВЂОпгавжжЦМЄЛюЕФGPЕФЙІФмЁЃЮЊСЫШЗБЃСзЫсЛЏ/ШЅСзЫсЛЏЗДгІЕФЬивьадЃЌGSКЭGPЖМПЩвдНсКЯЕНФмЙЛБЛСзЫсУИКЭМЄУИЪЖБ№ЕФЬЧдАаЯђЕААзЩЯЁЃетаЉЬЧдАаЯђЕААзОпгаВЛЭЌЕФзщжЏЗжВМКЭЖдGPЁЂGSКЭPP1ЕФВЛЭЌЧзКЭСІЁЃР§ШчЃЌБОЮФбаОПЕФгЩPpp1R3cЛљвђБрТыЕФPTGдкжЌЗОЯИАћЕШЖдвШЕКЫиУєИаЕФЯИАћжаБэДяЁЃгыИЮдрЛђМЁШтЯИАћЯрБШЃЌжЌЗОЯИАћжаЬЧдЫЎЦНЮЌГждкНЯЕЭЫЎЦНЃЌВЂБэЯжГіЖРЬиЕФЕїПиЁЃР§ШчЃЌжЎЧАдкДѓЪѓИЮдржаЕФбаОПНсЙћЯдЪОЃЌНћЪГ24аЁЪБКѓДѓЪѓИЮдрЦЯЬбЬЧ-6-СзЫсЫЎЦНЯТНЕСЫ5-6БЖЃЌЖјЕБИјЬЧФђВЁДѓЪѓзЂЩфвШЕКЫиЪБЃЌИЮЬЧдГСЛ§ЯджјдіМгЁЃЖјжЌЗОжаЕФЬЧдКЌСПдк48аЁЪБЕФНћЪГКѓЪмЕНСЫЯджјЕФвжжЦЃЌЖјдкЛжИДвћЪГКѓдђЯджјЕФМБадЩЯЕїЁЃЖдзиЩЋжЌЗОзщжЏжаЬЧдЕФбаОПжИГіЃЌдкНћЪГКѓЕФдйЮЙбјЃЈre-fedЃЉЦкМфЃЌBATЬЧддкжЌжЪВЙГфжЎЧАЗЂЩњЙ§ЖШЛ§РлЃЌВЂдкжЌжЪЛжИДКѓЬЧдЫЎЦНЛжИДЕНЛљЯпЫЎЦНЃЌИУЙ§ГЬЪмЕНЩіЩЯЯйЫиФмМЄЫиЕФбЯИёПижЦЁЃДЫЭтЃЌгабаОПжИГіЃЌжЌЗОЯИАћКЭОоЪЩЯИАћБэЯжГіЦЯЬбЬЧЧуЯђадЬЧдКЯГЩЁЃiWATЯИАћжаЬЧдЕФДѓСПЛ§РлПЩвдЭЈЙ§mTORC1вжжЦв§Ц№здЪЩЃЌЪЭЗХЪнЫиЃЌЮќв§M0ЦкЕФОоЪЩЯИАћМЄЛюзЊЯђM1ЦкЃЌЗжУкДйбзадЯИАћвђзгв§Ц№бзжЂЁЃетвВЗДЙ§РДгжжБНггАЯьжЌЗОЯИАћЕФЗжУкЙІФмЃЌдьГЩвШЕКЫиЕжПЙЁЃвдЩЯНсЙћЬсЪОжЌЗОжаЕФЬЧдЫЎЦНБфЛЏДцдкгыФмСПзДЬЌЯрЙиЕФМБадЕїНкзїгУЁЃ
ЕЋЪЧЃЌжБЕНЯждкжЌЗОжаЬЧдФмЙЛЦ№ЕНЪВУДбљЕФзїгУШдШЛЪЧИіУеЁЃНќЦкЗЂБэдкNatureдгжОЩЯЕФвЛЦЊЬтЮЊЁАGlycogen metabolism links glucosehomeostasis to thermogenesis in adipocytesЁБЕФЮФеТвдаТгБЕФНЧЖШНтД№СЫетИіУеЬтЃЌЯъЯИНщЩмСЫжЌЗОЯИАћжаЬЧдДњаЛКЭВњШШжЎМфЕФЙиЯЕЁЃ

БОбаОПЗЂЯжЃЌЬЧддкЕїНкiWAT(Inguinal white adipose tissueЃЌИЙЙЩЙЕАзЩЋжЌЗОЃЌИЛКЌУзЩЋжЌЗОЯИАћ)зиЩЋЛЏжаЦ№ЕНСЫЙиМќзїгУЁЃЬЧдЕФЛ§РлКЭНЕНтЃЌПЩвдЕїНкжЌЗОЯИАћЕФФмСПЯћКФКЭШШСПВњЩњЁЃЭЈЙ§МЄЛюНЛИаЩёОДЬМЄжЌЗОЯИАћВњШШЃЌЕМжТжЌЗОЯИАћжаЬЧдЕФЖЏЬЌЛ§РлКЭзЊЛЛЁЃетвЛЗЂЯжНЋЬЧДњаЛгыжЌЗОВњШШСЊЯЕЦ№РДЃЌгаРћгкДйНјДњаЛНЁПЕЕФИФЩЦЃЌЛђаэЛсЪЧЮДРДжЮСЦЗЪХжКЭЦфЫћДњаЛадМВВЁЕФаТЫМТЗЁЃ
ЧУКкАхРВЃЁ
1ЁЂЬЧддкУзЩЋжЌЗОжаИЛМЏЃЌЦфДњаЛПЩвдЕїПиUCP1ЕФБэДяЁЃ
2ЁЂІТ-ЩіЩЯЯйЫиЩЯЕїжЌЗОЯИАћжаЬЧдЕФКЯГЩКЭзЊЛЛЃЌгУвдЕїНкВњШШЁЃ
3ЁЂжЌЗОЯИАћЯИАћжЪЪЧЬЧдЖЏЬЌЕїНкROSВњЩњМАгеЕМp38МЄЛюЕФГЁЫљЁЃ
4ЁЂPTG-KOаЁЪѓдкГЄЦкРфБЉТЖКѓЃЌO2ЯћКФЁЂCO2ВњСПКЭЬхЮТОљНЕЕЭЁЃ
баОПНсЙћ
1ЁЂЬЧддкУзЩЋжЌЗОЯИАћжаБЛгеЕМ
габаОПжИГіЃЌжЌЗОзщжЏжаЬЧдЫЎЦНдкНћЪГЁЂНјЪГКЭжчвЙбЛЗЪБЛсЗЂЩњЯргІЕФБфЛЏЃЌетБэУїЫќПЩФмЪмЕНІТ-ЩіЩЯЯйЫиФмаХКХЕФЕїНкЁЃзїепМьВтСЫбЁдёадІТ3ЩіЩЯЯйЫиФмМЄЖЏМСCL-316,243ДІРэЕФаЁЪѓiWATжаЬЧдДњаЛЛљвђЕФБэДяЁЃНсЙћЗЂЯжЃЌCL-316,243ДІРэаЁЪѓiWAT 7ЬьКѓЃЌUcp1КЭDio2ЕФБэДяЯджјдіМг(ЭМS1a)ЃЌетБэУїiWATЗЂЩњСЫзиЩЋЛЏЃЌУзЩЋжЌЗОЯИАћЛ§РлЃЛЭЌЪБЃЌЬЧдДњаЛЛљвђШчЗжБ№БрТыmGS (Muscle glycogen synthaseЃЌМЁЬЧдКЯГЩУИ)КЭPTGЕФGys1КЭPpp1r3cЕФБэДявВЩ§ИпСЫЁЃГіКѕвтСЯЕФЪЧЃЌБрТыhGS(Liver glycogen synthaseЃЌИЮЬЧдКЯГЩУИ)КЭСзЫсЛЏУИЕФGys2КЭPyglЕФБэДявВЯджјдіМг (ЭМS1b)ЃЈаЁБрзЂЃКИЮЬЧдКЯГЩУИhGSжївЊдкИЮдржаБэДяЃЌдкЦфЫћзщжЏБэДяГЬЖШЕЭЁЃЧвЦфЫћзщжЏжївЊЭЈЙ§МЁЬЧдКЯГЩУИБэДяЁЃзїепШЯЮЊИЮдрбЧаЭЕФБэДяПЩФмНіЯогкУзЩЋКЭзиЩЋжЌЗОЯИАћЁЃзїепЭЦВтетжжЬивьадЩЯЕїПЩФмгыУзЩЋКЭзиЩЋжЌЗОЯИАћЕФЦЯЬбЬЧЩуШЁдіЧПЁЂЬЧдвжжЦНЕЕЭгыВњШШгаЙиЁЃЃЉЁЃДЫЭтЃЌзїепЗЂЯжforskolinЃЈЯймеЫсЛЗЛЏУИМЄЛюМСЃЉЩЯЕїСЫШЫдДжЌЗОЯИАћжаGys1ЁЂPyglКЭPpp1r3cЕФБэДяЫЎЦН(ЭМS2a)ЁЃгыWATЯрБШЃЌBATжаUcp1ЁЂDio2КЭДйНјЬЧдДњаЛЛљвђЕФЛљДЁБэДяЫЎЦНИќИпЃЌвђДЫCl-316,243ДІРэЕФаЇЙћВЂВЛЯджј(ЭМS2b)ЁЃ
ЭиеЙдФЖС
ЬЧдДњаЛУИ
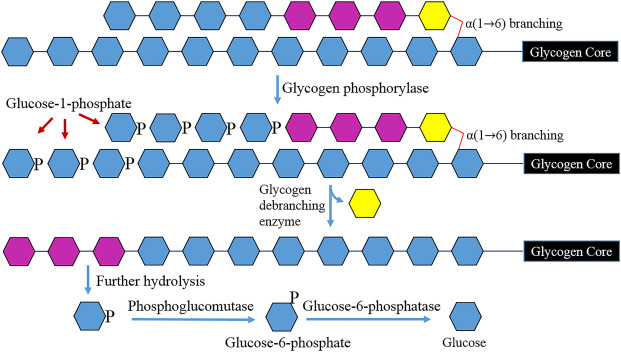
дкЬЧдКЯГЩЕФЙ§ГЬжаЃЌGS (Glycogen synthase, ЬЧдКЯГЩУИ)зїЮЊЕїПиКЯГЩЕФЙиМќУИЦ№ЕНСЫживЊЕФзїгУЁЃдкВИШщЖЏЮяЬхФкЃЌGSгЩGys1КЭGys2етСНИіЛљвђБрТыЃЌGys1жївЊдкМЁШтКЭЦфЫћзщжЏжаБэДя (MGS, Muscle glycogensynthase, МЁЬЧдКЯУИ)ЃЌЖјGys2дђжївЊдкИЮдржаЬивьадБэДя(H/LGS, Hepatic/Liver glycogen synthetase, ИЮЬЧдКЯУИ)ЁЃетСНжжЭЌЙЄУИжЎМфОпга70%ЕФађСаЯрЫЦЖШЃЌЕЋЫќУЧЕФбЧЯИАћЖЈЮЛШДгаКмДѓЕФВюБ№ЁЃЕБЯИАћФкЦЯЬбЬЧШБЗІЪБЃЌH/LGSжївЊМЏжадкЯИАћжЪжаЖјMGSЖЈЮЛгкЯИАћКЫЁЃЕБЦЯЬбЬЧЙЉгІГфзуКѓЃЌH/LGSЪмЕНЯьгІвЦЖЏЕНЬЧКЌСПИпЕФЯИАћФЄИННќЃЌMGSдђДгКЫжавзЮЛНјШыЯИАћжЪДпЛЏЬЧдКЯГЩЁЃ
дкЬЧдЗжНтЕФЙ§ГЬжаЃЌGP (Glycogen phosphorylase, ЬЧдСзЫсЛЏУИ)КЭGDE (Glycogen debranching enzyme, ЬЧдЭбжЇУИ)ЪЧжївЊЕФЙиМќУИЁЃGPдкЙЧїРМЁЁЂИЮдрКЭФдЕФЭЌЙЄУИЗжБ№ГЦЮЊGPMЁЂGPLКЭGPBЃЌБрТыЛљвђЗжБ№ЪЧPygmЁЂPyglКЭPygbЁЃЦфжаЃЌPyglЫљБрТыЕФИЮЬЧдСзЫсЛЏУИжївЊИКд№ТњзуШЋЩэЖдбЊЬЧЕФашЧѓЃЌЖјЦфгрСНепдђНідкзщжЏФкЬивьадБэДявдТњзуашвЊЁЃЕБЬЧдЗжНтПЊЪМЪБЃЌGPДпЛЏЬЧдЕФСзЫсЛЏзїгУЃЌЪЙЬЧдЗжзгДгЗЧЛЙдЖЫж№ИіЖЯПЊІС-1,4ЬЧмеМќвЦШЅЦЯЬбЬЧЛљЃЌЪЭЗХ1-СзЫсЦЯЬбЬЧЃЌжБжССйНќЬЧдЗжзгІС-1ЃЌ6-ЬЧмеМќЗжжЇЕуЧА4ИіЦЯЬбЬЧЛљДІЁЃжЎКѓгЩGPBМЬајЖЯПЊІС-1,4-ЬЧмеМќЃЌНЋИУЗжжЇЪЃгрЕФЧА3ИіЦЯЬбЬЧЛљвЦжССэвЛИіСэвЛЗжжЇФЉЖЫЃЌНЋЦфвдІС-1,4-ЬЧмеМќСЌНгЁЃШЛКѓЬЧдЭбжЇУИЪЙВаСєЕФІС-1,6-ЬЧмеМќСЌНгЕФЦЯЬбЬЧЫЎНтЃЌДгЖјЯћГ§ИУЗжжЇЃЌЪЭЗХвЛЗжзгЦЯЬбЬЧЁЃ

ВЮПМЮФЯзЃК
[1] Ferrer JC et al. FEBS Lett. 1997Oct 6;415(3):249-52.
[2] Villarroel-EspЈЊndola F et al. JCell Biochem. 2013 Jul;114(7):1653-64.
ЭиеЙдФЖС
ІТ-ЩіЩЯЯйЫиФмгыЬЧд
ЩіЩЯЯйЫиФмЪмЬхЪЧвЛРрНщЕМЖљВшЗгАЗЕїПизїгУЕФЪмЬхЃЌИљОнЦфЖдШЅМзЩіЩЯЯйЫиЕФВЛЭЌЗДгІЧщПіЃЌЗжЮЊЩіЩЯЯйЫиФмІСЪмЬхКЭІТЪмЬхЁЃ
дкЬЧдЕФКЯГЩгыЗжНтДњаЛжаЃЌІТ-ЩіЩЯЯйЫиФмЪмЬхЦ№ЕНСЫживЊЕФЕїПизїгУЁЃбаОПБэУїЃЌЕБЩіЩЯЯйЫиМЄЛюІТ-ЩіЩЯЯйЫиФмЪмЬхКѓЃЌЯймеЫсЛЗЛЏУИБЛМЄЛюЃЌcAMPЫЎЦНЩЯЕїЃЌв§Ц№ЬЧдКЯГЩЫЎЦННЕЕЭЃЌДйНјЬЧдЗжНтЙ§ГЬЁЃЕЋСэвЛЗНУцЃЌвВгабаОПБэУїЃЌІТ-ЩіЩЯЯйЫиФмМЄЛюПЩФмЛсЭЈЙ§МЄЛюPI3KЯТгЮЭЈТЗЕМжТаФМЁЯИАћКЭжЌЗОЯИАћжаGSK3(Glycogen synthase kinase 3,ЬЧдКЯГЩУИМЄУИ-3 )ЕФСзЫсЛЏЃЌДгЖјЕїПиЬхФкGSЕФСзЫсЛЏЫЎЦНЃЌЩЯЕїЬЧвьЩњЭООЖЃЌдіМгЬхФкЬЧдЛ§РлЁЃ
ВЮПМЮФЯзЃК
[1] Carmean CM et al. PLoS One. 2013Jul 4;8(7):e67807.
[2] Yamamoto DL et al. Diabetologia.2007 Jan;50(1):158-67.
дкWATжаЃЌвРРЕCL-316,243ЕФЬЧдДњаЛЛљвђЕФmRNAЫЎЦНЩ§ИпЃЌЭЌЪБmGSЁЂhGSЁЂhGPЕФЕААзЫЎЦНвВЩ§Ип(ЭМS1cЁЂS2c)ЁЃЫфШЛдкBATжаУЛгаМьВтЕНетаЉЕААзБэДяЕФВювьЃЌЕЋhGSЯдЪОГіНЯИпЕФЛљДЁБэДяЫЎЦНЃЌетгыВњШШЙІФмЕФЩЯЩ§гаЙиЃЈаЁБрзЂЃКгабаОПБЈЕРжИГіЃЌBATдкЧУГ§TGКЯГЩУИDGAT1КЭDGAT2ДгЖјНЕЕЭжЌЗОЫсДЂДцЕФЧщПіЯТЃЌПЩвдРћгУбЛЗЦЯЬбЬЧКЭжЌЗОЫсвдМАДЂДцЕФЬЧдРДДйНјВњШШЃЌвђДЫШЯЮЊЬЧдКЯГЩУИПЩвдЬсИпЬЧдКЌСПЃЌЯдЪОГігыВњШШЕФЯрЙиадЃЉ(ЭМS2d)ЁЃЪЙгУCL-316,243ДІРэЕФаЁЪѓжЌЗОЯИАћжаЕФЬЧдЫЎЦНДѓдМдіМгСЫ3БЖ(ЭМS1d)ЁЃUCP1жївЊдкЬЧдЫЎЦНзюИпЕФЖрЗПжЌЗОЯИАћжаБэДя(ЭМS1e)ЁЃЕчОЕЗжЮіЯдЪОЃЌдкЪЙгУCL-316,243ДІРэЕФаЁЪѓжЌЗОЯИАћжаЃЌдкЯпСЃЬхХдЃЈjuxtaposed to mitochondriaЃЉЕФЬЧдПХСЃИќЖр(ЭМS1f)ЁЃДгiWATжаЗжРыЕФГЩЪьжЌЗОЯИАћЗжЮівВжЄЪЕЃЌЩЯЪіЛљвђБэДяБфЛЏОпгажЌЗОЯИАћЬивьад(ЭМS1g)ЁЃ
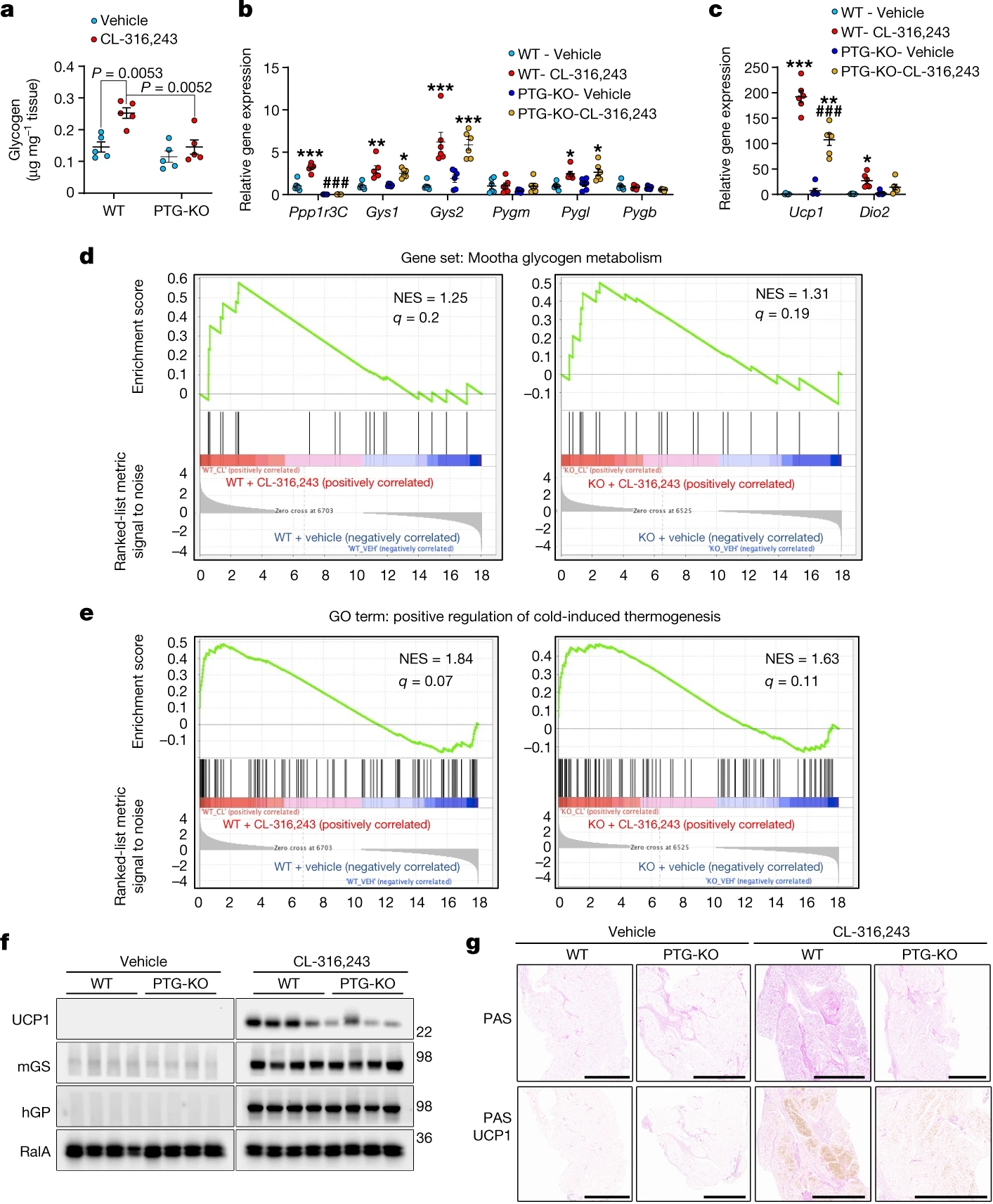
ЭМ1.PTG-KOНЕЕЭСЫУзЩЋжЌЗОЯИАћжаUCP1ЕФБэДя
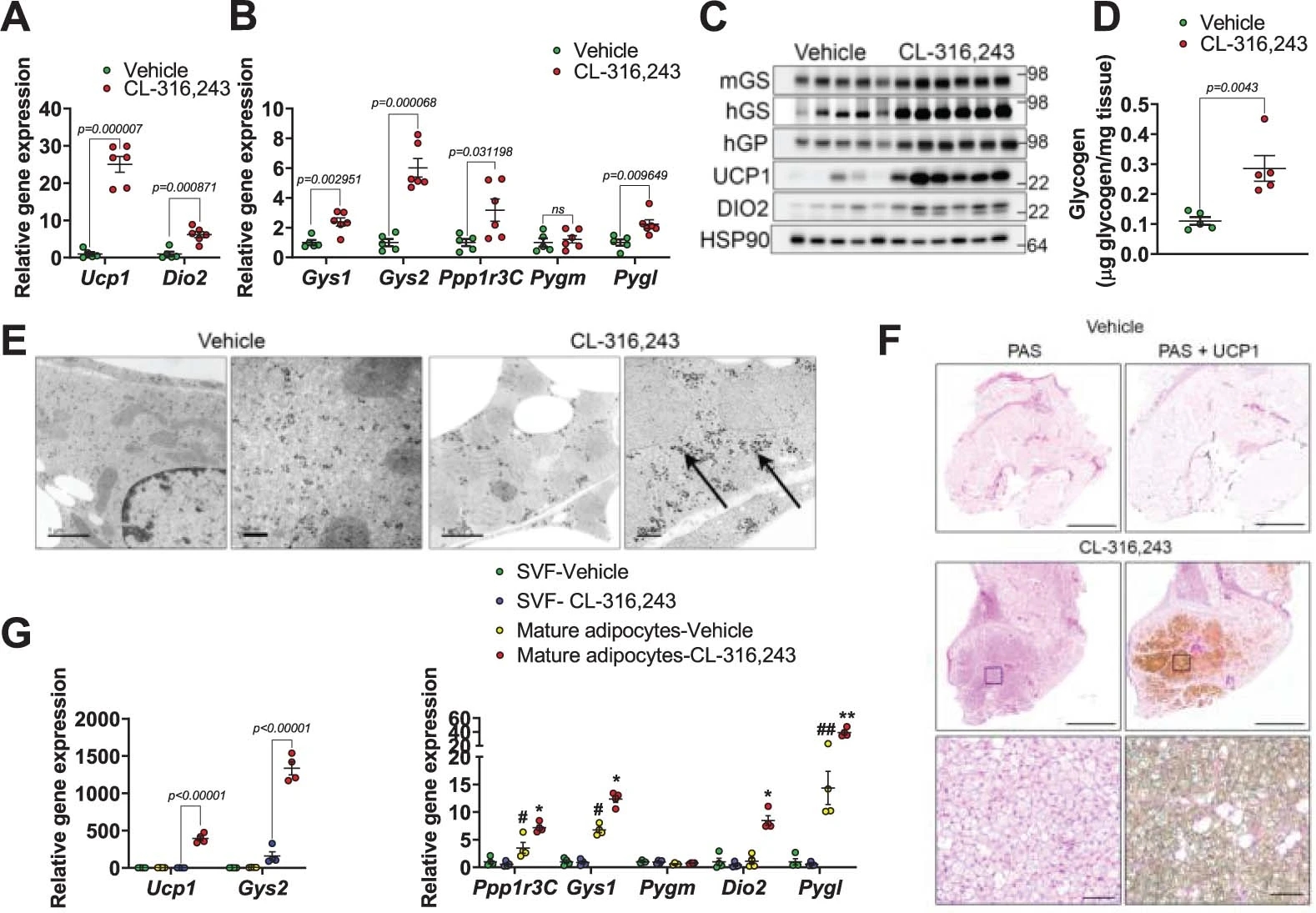
ЭМS1.ЬЧдДњаЛдкУзЩЋжЌЗОЯИАћжадіЧП
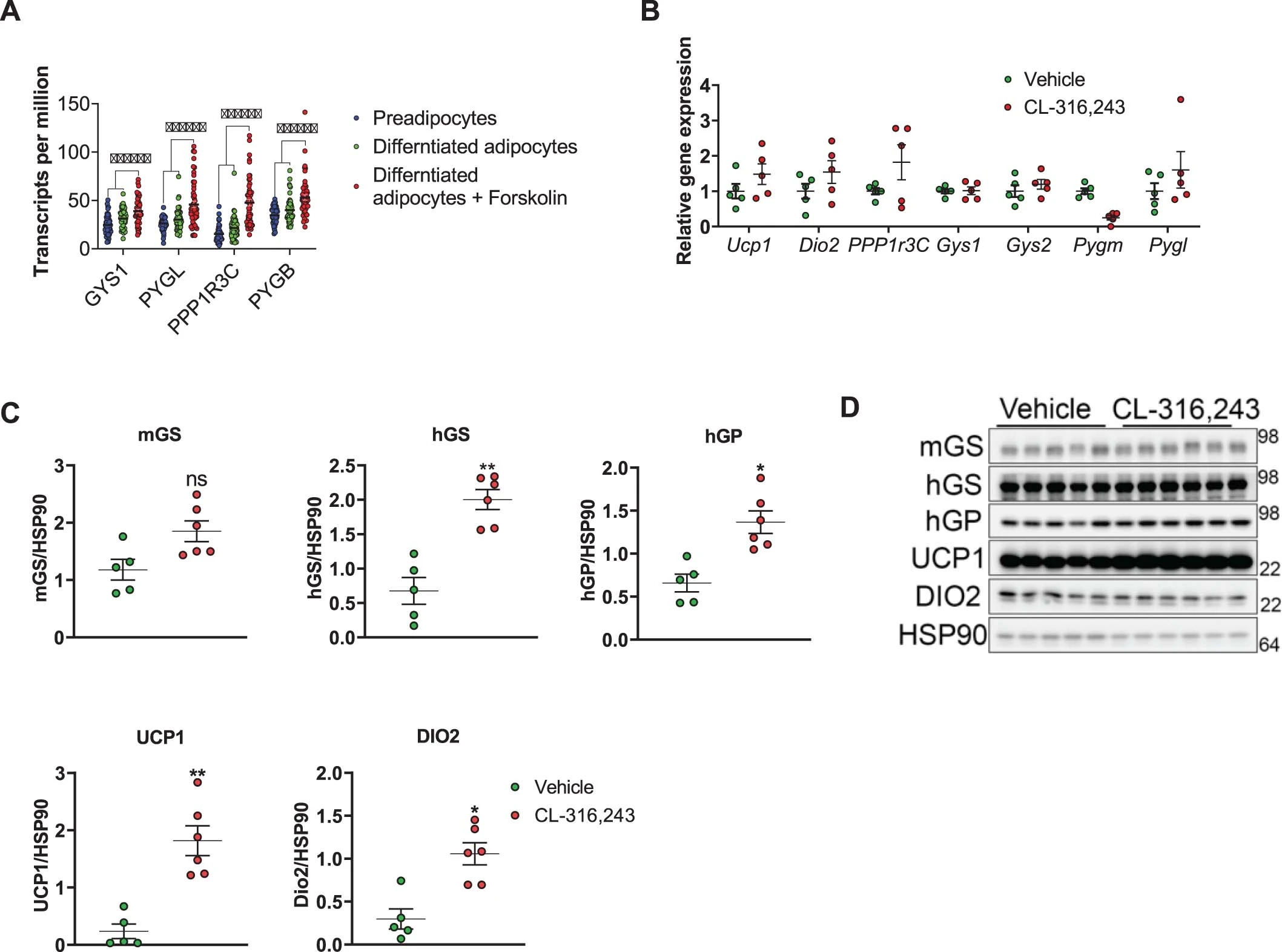
ЭМS2.CL-316,243 ДІРэВЛгАЯь BAT жаЬЧдДњаЛЛљвђЕФБэДя
2ЁЂЬЧдгАЯьUCP1ЕФБэДя
УзЩЋжЌЗОЯИАћжаЬЧдЛљвђБэДядіМгКЭЬЧдЛ§РлБэУїЦфгыВњШШЯрЙиЁЃАаЯђЧУГ§Ppp1r3cЕФPTG-KOаЁЪѓдкCL-316,243ДІРэКѓЬЧдЛ§РлМѕЩй(ЭМ1a)ЃЌЫцКѓUcp1КЭDio2ЕФБэДядіМгГЬЖШНЕЕЭ(ЭМ1c)ЁЃЖдRNA-seqНсЙћЕФВювьБэДяЗжЮівВжЄЪЕСЫетвЛЯжЯѓ(ЭМS3c)ЁЃГ§БрТыAgl(Glycogen debranching enzymeЃЌЬЧдШЅЗжжЇУИ)ЕФЛљвђЭтЃЌЬЧдДњаЛЛљвђдкВЛЭЌЛљвђаЭМфБэДяЮоЯджјВювь (ЭМS3c)ЁЃ
дкОCL-316,243ДІРэЕФWTКЭPTG-KOаЁЪѓЕФЗжРыЕФжЌЗОЯИАћжаЃЌGSEA(Gene set enrichment analysisЃЌЛљвђМЏИЛМЏЗжЮі)ЯдЪОPPAR (Peroxisome proliferator-activatedreceptorЃЌЙ§бѕЛЏЮяУИЬхдіжГЮяМЄЛюЪмЬх)аХКХЭЈТЗКЭЯпСЃЬхЩњЮяЗЂЩњЭООЖЕФЛљвђБэДяОљдіМг (ЭМS3d, S3e(аЁБрзЂЃКдЮФЪ§ОнS3 D-EЭМЦЌЮДЯдЪО))ЃЌетБэУїетаЉЭООЖВЛЪмЬЧдЫЎЦНЕФгАЯьЁЃЯрБШжЎЯТЃЌGOЗжЮіЯдЪОЃЌРфДЬМЄгеЕМВњШШЕФЁАе§ЕїПиЁБЭЈТЗЃЌдкWTжЌЗОЯИАћжаИЛМЏЃЌЖјдкPTG-KOаЁЪѓжаУЛгаБЛИЛМЏЃЌБэУїЬЧдЪЧВњШШЛљвђБэДяЕФЙиМќЕїПивђзг(ЭМ1d)ЁЃДЫЭтЃЌОCL-316,243ДІРэКѓЃЌWTКЭPTG-KOаЁЪѓЕФжЌЗОЯИАћжаЬЧдДњаЛЭООЖЯрЙиЛљвђОљгаЫљИЛМЏ (ЭМ1e)ЁЃ
дкPTG-KOаЁЪѓЕФiWATжаUCP1ЕААзБэДяНЕЕЭ(ЭМ1fЃЌS2f)ЁЃiWATзщжЏбЇНсЙћЯдЪОЃЌCL-316,243геЕМЕФЬЧдЫЎЦНКЭUCP1БэДяОљЯджјНЕЕЭ(ЭМ1g)ЁЃUCP1БэДяНЕЕЭгыЖдCL-316,243ЕФУєИаадЯТНЕЮоЙиЃЌвђЮЊМЄЫиДЬМЄКЭЛљДЁжЌНтдкВЛЭЌЛљвђаЭаЁЪѓжЎМфЕФЧщПіЯрЫЦ(ЭМS3g)ЁЃ
ЮЊСЫХХГ§ШЋЩэЧУГ§ЕФгАЯьЃЌзїепЙЙНЈСЫPTGжЌЗОзщжЏЬивьадЧУГ§аЁЪѓ(PTG-AKOаЁЪѓ)ЁЃдкPTG-AKOаЁЪѓЕФiWATжаЃЌCL-316,243геЕМЕФUCP1БэДяЯджјНЕЕЭЃЌетжЄЪЕСЫжЌЗОЯИАћЬЧдДњаЛЖдUCP1БэДяЕФжБНггАЯь(ЭМ2aЃЌЭМS3a)ЁЃдкCL-316,243ДІРэЕФЧА3ЬьЃЌВЛЭЌЛљвђаЭаЁЪѓжЎМфЕФКФбѕСПЯрЫЦ(ЭМ2b)ЁЃЕЋЫцКѓЕФМИЬьРяЃЌCl-316,243ДІРэЕФWTЪѓЕФКФбѕСПдіМгЃЌЖјPTG-AKOаЁЪѓдђУЛга(ЭМ2c)ЁЃCO2ВњЩњСПвВгаРрЫЦЕФЧїЪЦ(ЭМS4b, c)ЁЃPTG-AKOаЁЪѓгыWTаЁЪѓдкИпжЌвћЪГгеЕМЯТЕФЬхжиВЂУЛгаВювь(ЭМ2d)ЁЃДЫЭтЃЌгыЖдеезщЯрБШЃЌCL-316,243ДІРэPTG-AKOаЁЪѓЕМжТUCP1геЕМНЕЕЭ(ЭМ2eЃЌS3d-e)ЁЃВЛНіШчДЫЃЌдкPTG-AKOаЁЪѓжаЃЌгЩгкФмСПЯћКФНЕЕЭЃЌCL-316,243ЕМжТЕФЬхжиМѕЧсУїЯдИќЩй(ЭМ2fЃЌS3f-g)ЁЃ
змЕФРДЫЕЃЌетаЉЪ§ОнБэУїЬЧдДњаЛЪЧе§ГЃКЭЗЪХжаЁЪѓЪЪгІадВњШШЫљБиашЕФЁЃЮЊСЫШЗЖЈгыШЫРрЕФЯрЙиадЃЌзїепЖдРДздЗЧЗЪХж(n = 26)КЭЗЪХж(n = 30)ХЎадЦЄЯТжЌЗОЛюМьЕФЮЂеѓСаЪ§ОнНјааСЫЯпадЛиЙщЗжЮіЁЃGys2ЁЂPygmЁЂAglЕФБэДяЫЎЦНгыBMI(ЬхжижИЪ§)ГЪИКЯрЙиЁЃВЛНіШчДЫЃЌGys2ЁЂPygmЁЂPpp1r3dЕФБэДяЫЎЦНгыlog10 HOMA-IRЃЈвШЕКЫиЕжПЙЮШЬЌФЃаЭЦРЙРЃЉвВГЪИКЯрЙи(ЭМ2h)ЁЃЕкЖўзщАќКЌ770УћФаадЕФЖгСабщжЄСЫетаЉНсЙћЁЃЦфжаЃЌGys2ЁЂPygmгыBMI ЁЂHOMA-IR КЭбќЭЮБШГЪИКЯрЙи(ЭМS4h)ЁЃзмжЎЃЌдквдадБ№ЛЎЗжЕФСНзщЖРСЂЕФШЫРрЪмЪдепжаЃЌжЌЗОзщжЏжаЬЧдДњаЛЛљвђЕФИпБэДягыНЯЕЭЕФЬхжиКЭНЯИпЕФвШЕКЫиУєИаадЯрЙиЁЃ
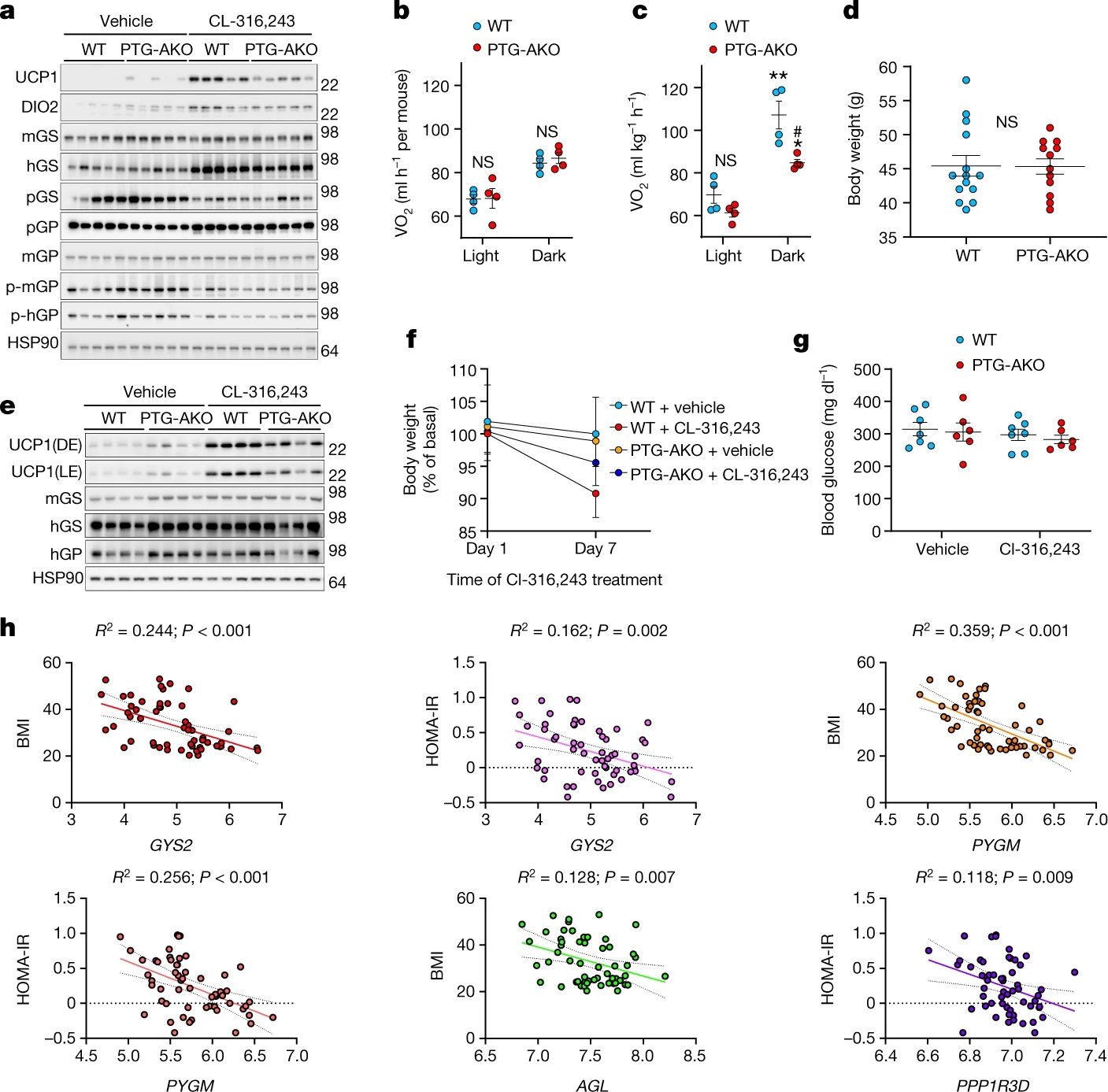
ЭМ2.жЌЗОЬивьад PTG ЧУГ§МѕЩйСЫ UCP1 БэДяКЭФмСПЯћКФ
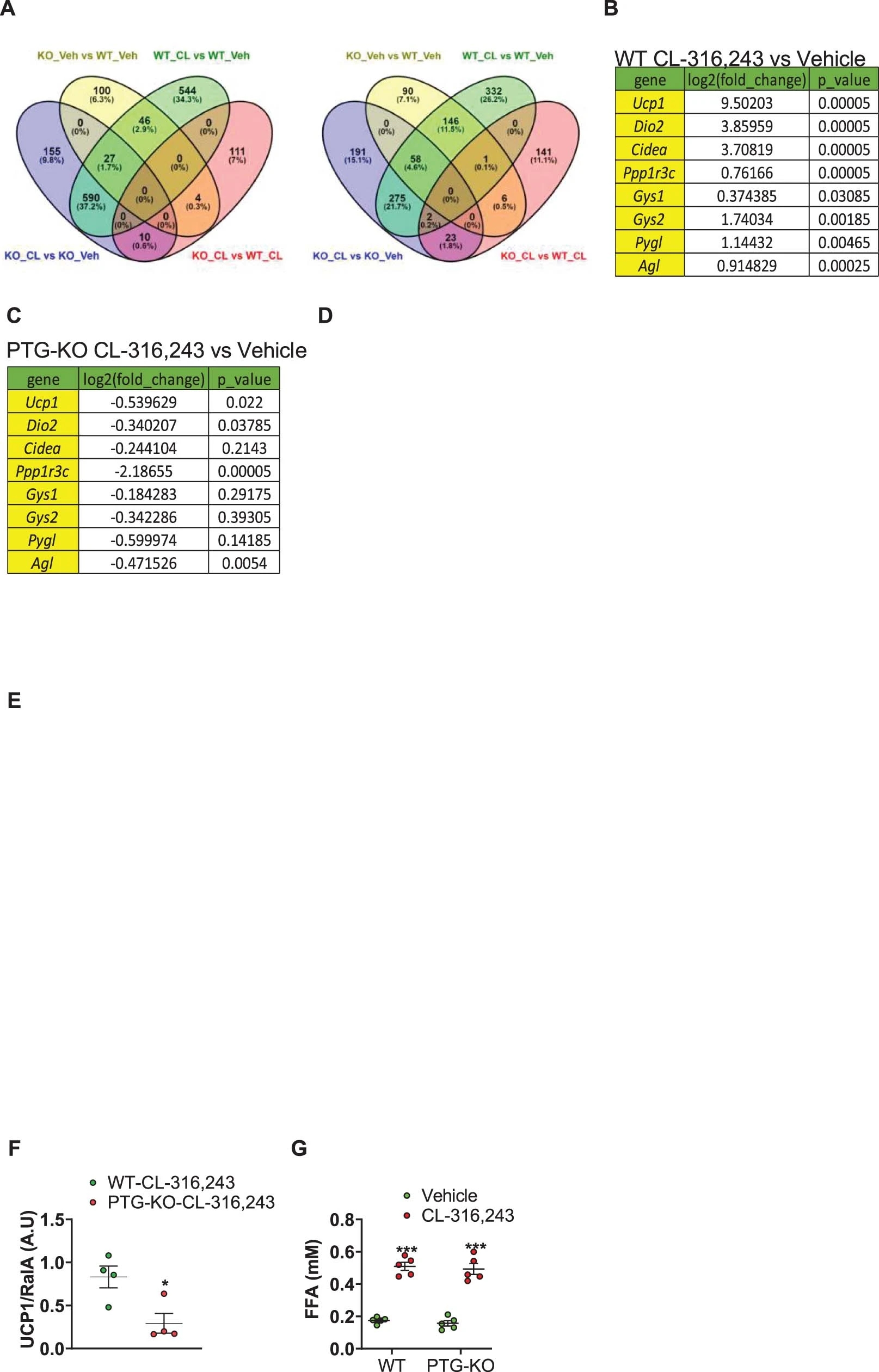
ЭМS3.PTG-KO ВЛгАЯьЖд CL-316,243 ЕФЯьгІ
ЃЈаЁБрзЂЃКS3 D-EЭМЦЌФПЧАдкЙйЭјДІгкЭМЦЌШБЪЇзДЬЌЃЌЖдгІЭјжЗЮЊhttps://www.nature.com/articles/s41586-021-04019-8/figures/7)
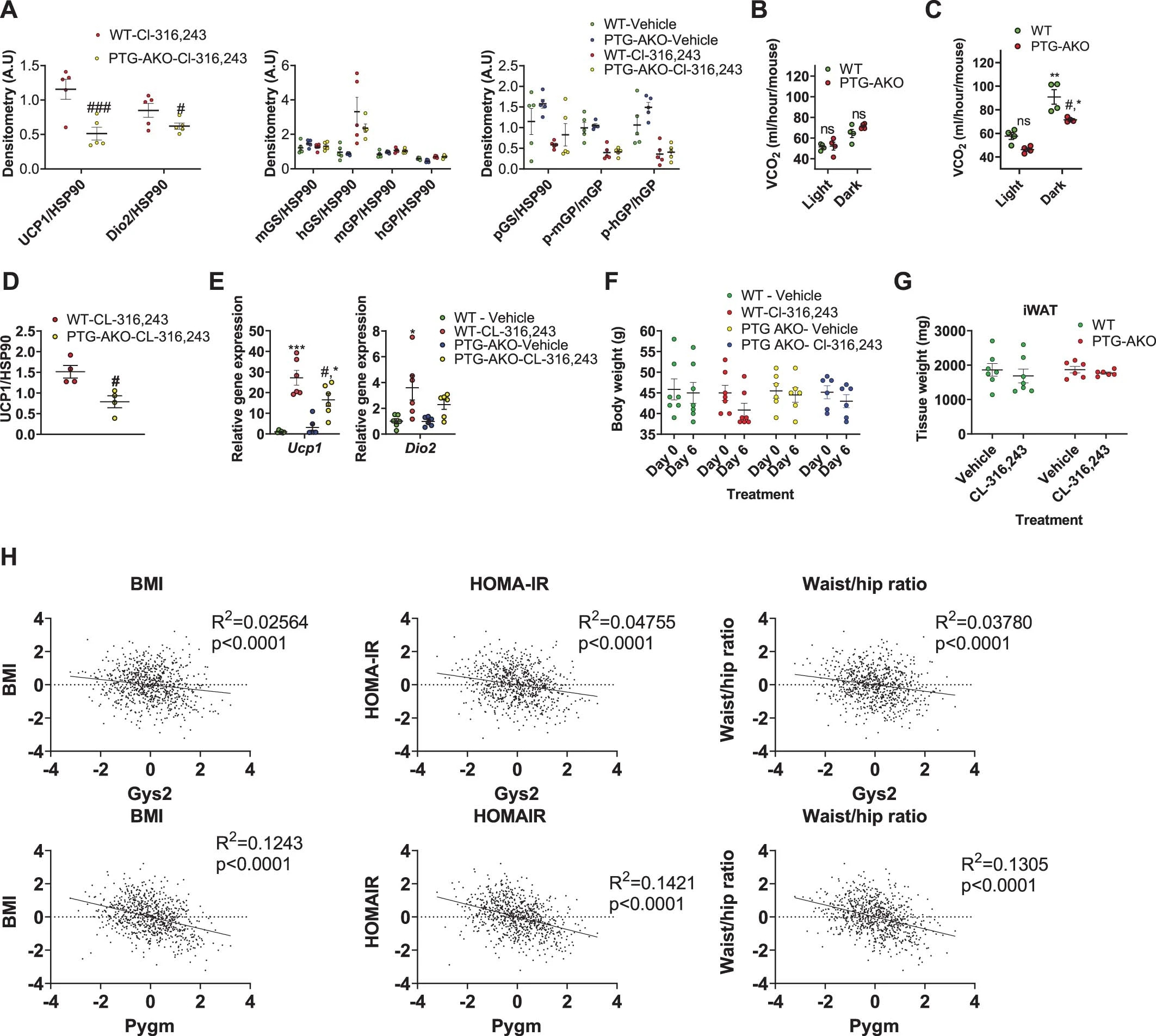
ЭМS4.жЌЗОЬивьадЧУГ§ PTG НЕЕЭСЫФмСПЯћКФ
3ЁЂЬЧдгАЯьp38 MAPKМЄЛю
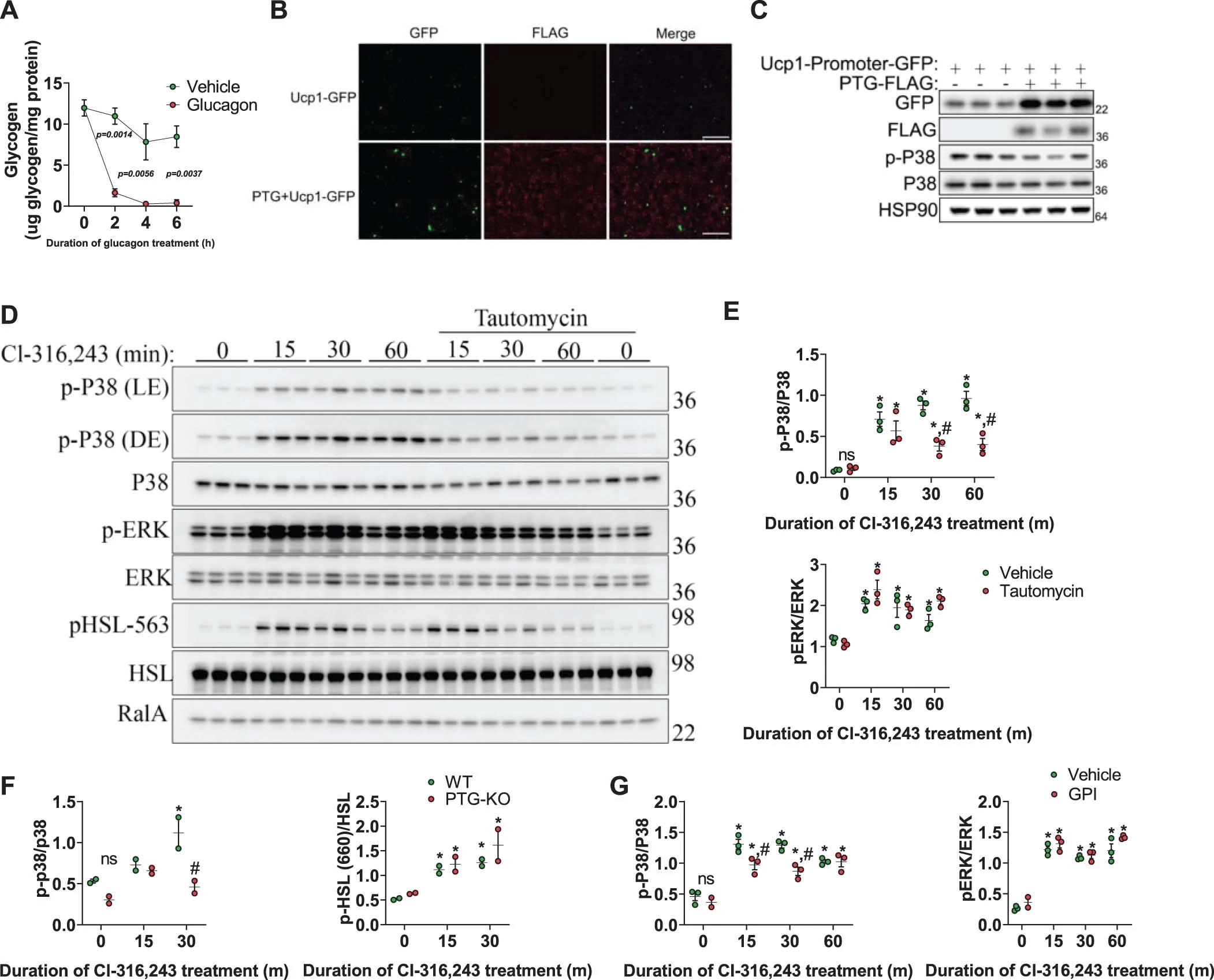
ЮЊСЫНјвЛВНЬНОПЬЧдЯьгІІТ3-ЩіЩЯЯйЫиФмМЄЛюЕФЖЏСІбЇЛњжЦЃЌзїепЪЙгУCL-316,243ДІРэЬхЭтЗжЛЏЕФiWATдДњЧАЬхжЌЗОЯИАћЃЌНсЙћЗЂЯжРДдДWTаЁЪѓЕФЧАЬхжЌЗОЯИАћжаЬЧдДѓСПЛ§РлЃЌЖјPTG-KOаЁЪѓдђУЛга(ЭМ3a)ЁЃДЫЭтЃЌдкPTG-KOжЌЗОЯИАћжаЃЌЬЧдЗжНтЕФжеВњЮяG1P(Glucose-1-phosphateЃЌЦЯЬбЬЧ-1-СзЫс)ЕФЫЎЦНвВДѓЗљНЕЕЭ(ЭМ3b)ЃЌЖјвШИпбЊЬЧЫиДІРэPTG-KOаЁЪѓЕФИЮдДњЯИАћдђПЩНЕЕЭЦфжаЕФЬЧдЫЎЦНЃЌБэУїИЮЯИАћЕФЬЧдЗжНтВЛЪмгАЯь(ЭМS5a)ЃЌЫЕУїетСНжжЯИАћРраЭжЎМфЕФЗДгІДцдкВювьЁЃCL-316,243ДЬМЄPTG-KOЗжЛЏЕФжЌЗОЯИАћЯдЪОUcp1КЭDio2ЕФБэДяМѕЩй(ЭМ3c)ЁЃДЫЭтЃЌдкЙВБэДяPTGЕФЯИАћжаЃЌUcp1ЦєЖЏзгЕїПиЕФGFPБэДяЯджјдіМг(ЭМS5b, c)ЃЌБэУїЖљВшЗгАЗдіМгСЫжЌЗОЯИАћжаЬЧдЕФКЯГЩКЭзЊЛЛЃЌНјЖјЕїНкВњШШЁЃ
вжжЦPKAЛђp38ПЩвдМѕШѕCL-316,243геЕМЕФWTжЌЗОЯИАћ(ЭМ3d)КЭPTG-KOжЌЗОЯИАћжаUcp1ЕФБэДяЁЃжЕЕУзЂвтЕФЪЧЃЌCL-316,243ДІРэКѓЃЌPTG-KOжЌЗОЯИАћжаCL-316,243вРРЕЕФp38СзЫсЛЏМЄЛюЫЎЦНБЛЯТЕїЃЌЖјвРРЕгкPKAЕФHSL(Hormone sensitive lipaseЃЌМЄЫиУєИааджЌЗОУИ)СзЫсЛЏдђЮДЪмгАЯь(ЭМ3eЃЌЭМS5f)ЁЃгыPTG-KOжЌЗОЯИАћРрЫЦЃЌЪЙгУБфЙЙУЙЫиЃЈаЁБрзЂЃКБфЙЙУЙЫиЪЧвЛжжЕААзСзЫсУИЬивьадвжжЦМСЃЌЖдPP1КЭPP2AОљгааЇЃЌЦфжаЖдPP1ЕФвжжЦаЇЙћЧПгкPP2AЃЌвђДЫБЛЙуЗКгУгквжжЦPP1ЁЃЃЉвжжЦPP1ПЩНЕЕЭЖљВшЗгАЗДЬМЄЯТp38ЕФСзЫсЛЏЫЎЦН(ЭМS5d, e)ЃЌетаЉНсЙћБэУїЬЧдПЩвдЖЏЬЌЕїНкp38ЕФМЄЛювдМАЫцКѓЕФUCP1ЕФгеЕМЁЃ
гЩгкGSКЭGPЕФБэДяЖМЪЧдкCL-316,243ДЬМЄгеЕМЕФЃЌзїепЭЦВтЬЧдзЊЛЛПЩвдЕїНкUCP1ЕФБэДяЁЃвђДЫЃЌзїепЪЙгУGPI(GP inhibitorЃЌGPвжжЦМС)зшЖЯЬЧдЗжНтЃЌЗЂЯжGPIдіЧПСЫІТ3-ЩіЩЯЯйЫиФмМЄЛюКѓЬЧдЕФЛ§Рл(ЭМ3f)ЃЌЕЋНЕЕЭСЫp38ЕФМЄЛю(ЭМ3gЃЌЭМS5g)ЃЌВЂЫцКѓНЕЕЭСЫUcp1КЭDio2ЕФБэДя(ЭМ3h)ЁЃетБэУїЬЧдЕФзЊЛЛЪЧжЌЗОЯИАћжаp38МЄЛюЫљБиашЕФЁЃ
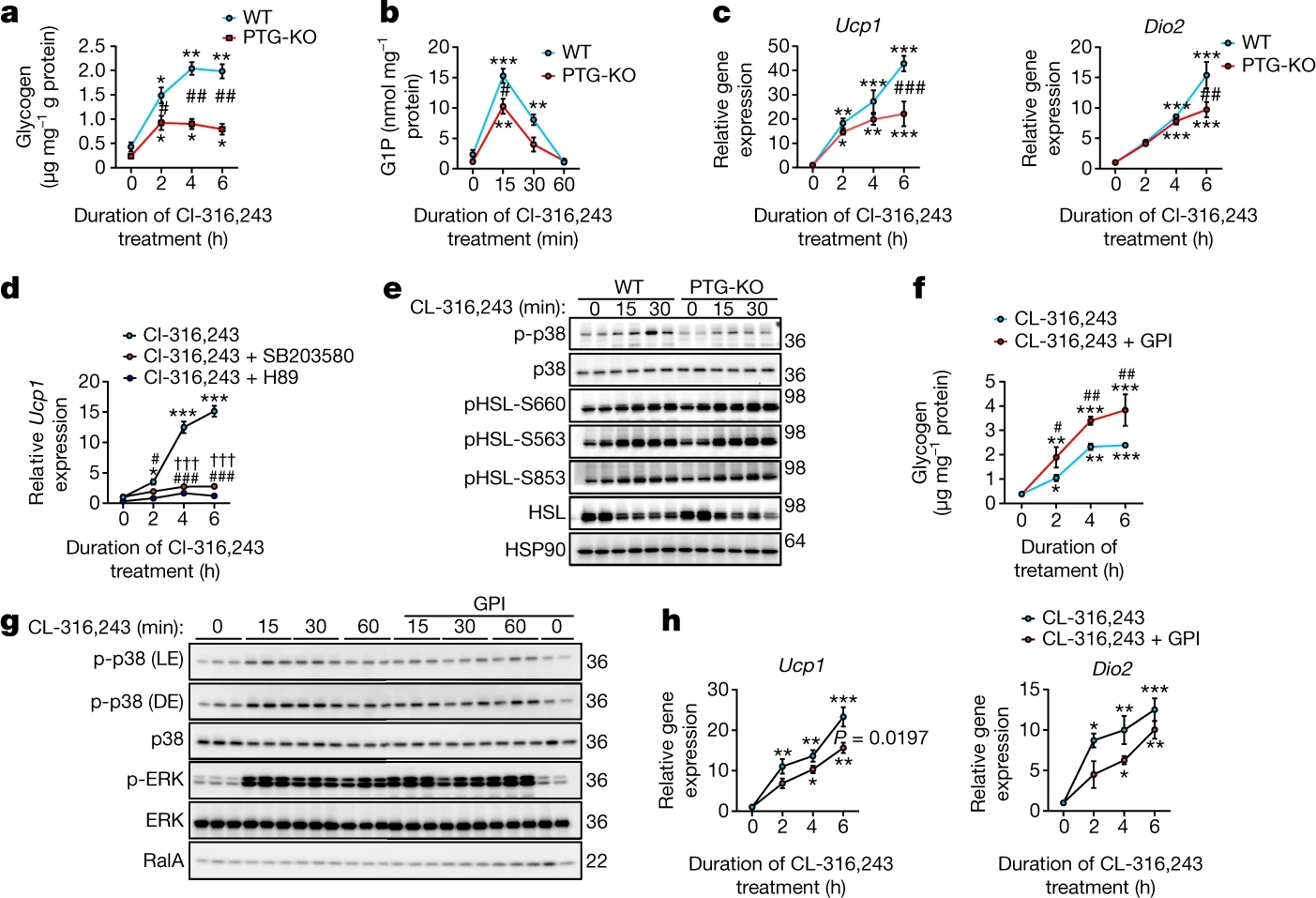
ЭМ3.ЬЧдДњаЛЕФЯТЕївжжЦСЫ p38 ЕФМЄЛю
ЭМS5.PTG ЕФЙ§БэДяЩЯЕїСЫЬхЭтUCP1 ЕФБэДя
4ЁЂЬЧдЕїНкROSвРРЕЕФp38МЄЛю
гЩгкROSЕФВњЩњЪЧМЄЛюp38ЫљБиашЕФЃЌвђДЫзїепЪЙгУCL-316,243ДЬМЄЗжЛЏЕФжЌЗОЯИАћЗЂЯжЃЌROSЫЎЦНЯджјдіМг(ЭМ4a)ЃЌПЙбѕЛЏЛљвђБэДявВЯргІдіМгЃЌЬэМгROSЧхГ§МСNAC(N-Acetyl-L-cysteineЃЌN-ввѕЃ-L-АыызАБЫс)КѓЃЌПЩвдНЕЕЭетаЉЛљвђЕФБэДя(ЭМ4b)ЁЃЭЌЪБЃЌNACФмЙЛЯджјНЕЕЭCL-316,243геЕМЕФp38МЄЛювдМАUcp1КЭDio2ЕФБэДя(ЭМ4c, dЃЌЭМS6a)ЃЌетБэУїp38ЕФМЄЛюКЭЫцКѓUCP1БэДяЕФдіМгвРРЕгкROSЕФВњЩњЁЃ
ЭиеЙдФЖС
ЬЧдЁЂp38гыROS
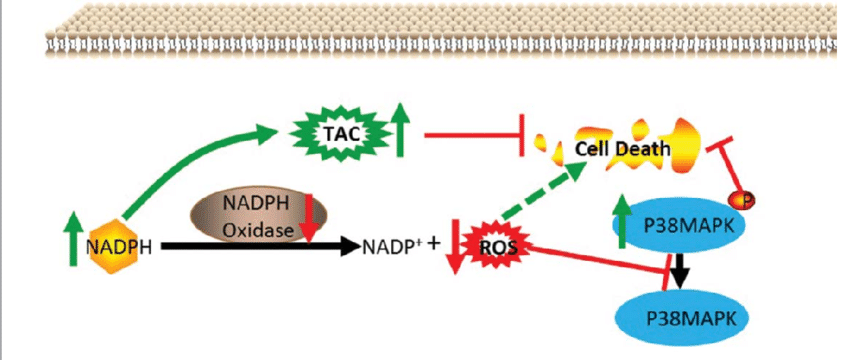
ИУбаОПШЯЮЊЦЯЬбЬЧбѕЛЏЪЧЯИАћжЪжаROSЕФжївЊРДдДЃЌвђЖјЭЦВтдкЬЧдЗжНтжабмЩњЕФ1-СзЫсЦЯЬбЬЧЮЊЬЧНЭНтЬсЙЉСЫЕзЮяЁЃЖјЬхФкЬЧНЭНтЛсв§Ц№NADPHЕФДѓСПВњЩњЃЌВЂЭЈЙ§дкжЪФЄКЭФкжЪЭјЩЯБэДяЕФNOX(NADPH oxidase, NADPHбѕЛЏУИ)НЋNADPHжаЕФЕчзгзЊвЦЕНбѕЩЯЃЌВњЩњДѓСПROSЁЃВњЩњЕФROSгжПЩвдЕїПиЬхФкp38ЕФБэДяЁЃ
габаОПБэУїЃЌдкЩњРэЬѕМўЯТЃЌp38ПЩвдзїЮЊROSаХКХзЊЕМЕФНщжЪЃЌВЂИљОнВЛЭЌДЬМЄЃЌМЄЛюЛђвжжЦЯИАћжмЦкНјГЬЁЃЕБЯИАћДІгкШБбѕЬѕМўЯТЃЌЯИАћФкВњЩњДѓСПROSЃЌДгЖјДйНјp38ЕФСзЫсЛЏЁЃетжжСзЫсЛЏМДЪЙдкЯИАћзДЬЌЛжИДКѓШдЛсМЬајЃЌвђЮЊp38ЕФСзЫсЛЏМЄЛюгжЛсНјвЛВНгАЯьROSЕФВњЩњЃЌДгЖјЯнШыЖёадбЛЗЁЃдкBATжаЃЌСзЫсЛЏКѓЕФp38ПЩвдЭЈЙ§ATF2(Activating transcription factor 2,МЄЛюзЊТМвђзг2)ЃЌЪЙЦфзЊдЫжСЯИАћКЫВЂНсКЯдкUcp1КЭPgc1ІСЕФЦєЖЏзгЩЯЃЌДгЖјЕїПиUcp1КЭPgc1ІСБэДяЃЌгАЯьВњШШЁЃ
ВЮПМЮФЯзЃК
[1]Tormos AM et al. Free Radic Res. 2013 Nov;47(11):905-16.
[2]Fischer K et al. Nat Commun. 2020 May 8;11(1):2306.
[3]Bedard K, Krause KH. Physiol Rev. 2007 Jan;87(1):245-313.
[4] Pei H et al. Cell Cycle. 2017 Jan 2;16(1):113-122.
RNA-seqЪ§ОнЕФGSEAЯдЪОЃЌЁАROSПЙбѕЛЏЗДгІЁБЭЈТЗдкCL-316,243ДІРэЕФWTаЁЪѓЕФжЌЗОЯИАћжаИпЖШИЛМЏЃЌЖјдкPTG-KOжЌЗОЯИАћжадђЭъШЋШБЪЇ(ЭМ4e)ЁЃPTGЧУГ§ЯджјНЕЕЭСЫCL-316,243в§Ц№ЕФROSВњЩњ(ЭМ4f)ЃЌЧввжжЦGPвВФмЯджјНЕЕЭROSЃЌетБэУїЬЧдЕФзЊЛЛЪЧВњЩњROSЫљБиашЕФЁЃЖдМгШыCL-316,243ЛђCL-316,243+GPIДІРэЕФжЌЗОЯИАћЕФАыызАБЫсбѕЛЏНјааећЬхЗжЮіЗЂЯжЃЌдкМгШыGPIЕФІТ-ЩіЩЯЯйЫиФмДЬМЄКѓЃЌЬиЖЈЕААзбЧШКжаВЛЭЌЕААзЕФАыызАБЫсбѕЛЏзДЬЌЃЈаЁБрзЂЃКгЩгкROSПЩвдЭЈЙ§гАЯьФкЛЗОГбѕЛЏЛЙдЫЎЦНЃЌДгЖјгАЯьЕААзжЪАыызАБЫсбѕЛЏЛЙдаоЪЮЫЎЦНЁЃвђЖјзїепРћгУuniprotбЁЖЈСЫЪмАыызАБЫсбѕЛЏЕїПиЕФЕААзбЧШКЁЃЭЈЙ§ЖдАыызАБЫсбѕЛЏЕїПиЕФМьВтПЩвдбаОПЯИАћФкЕААзЕФбѕЛЏЛЙдЧщПіЃЌИУНсЙћБэУїЃЌЕБвжжЦGPКѓЃЌЯИАћФкЕААзЕФбѕЛЏЛЙдЫЎЦНЗЂЩњИФБфЃЌДгЖјЭЦЖЯГіROSЪмGPЕїПиЕФЬЧдДњаЛЕїНкЃЉЗЂЩњСЫБфЛЏ(ЭМS6b, c)ЁЃЪЙгУЯпСЃЬхROSЬНеыЃЈCM-H2DCFDAЃЉНјвЛВНбаОПЗЂЯжЃЌдкCL-316,243ДІРэжЌЗОЯИАћКѓЃЌЯпСЃЬхROSдіМгЃЌЕЋгыGPЮоЙи(ЭМ4g)ЁЃвђДЫЃЌЬЧдЕФКЯГЩКЭЗжНтЕМжТROSЕФВњЩњКЭp38ЕФМЄЛюКмПЩФмЗЂЩњдкЯИАћжЪжаЁЃ
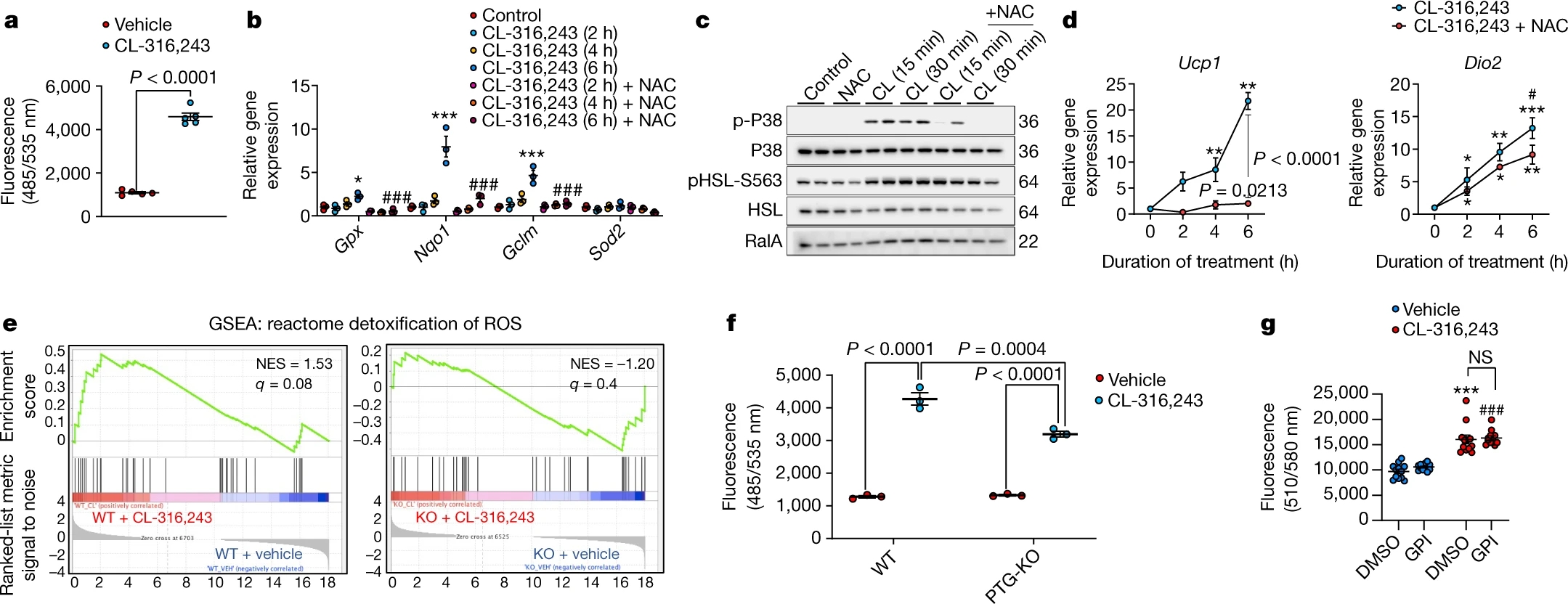
ЭМ4. ЬЧдДњаЛДйНјжЌЗОЯИАћжа ROS ЕФВњЩњ
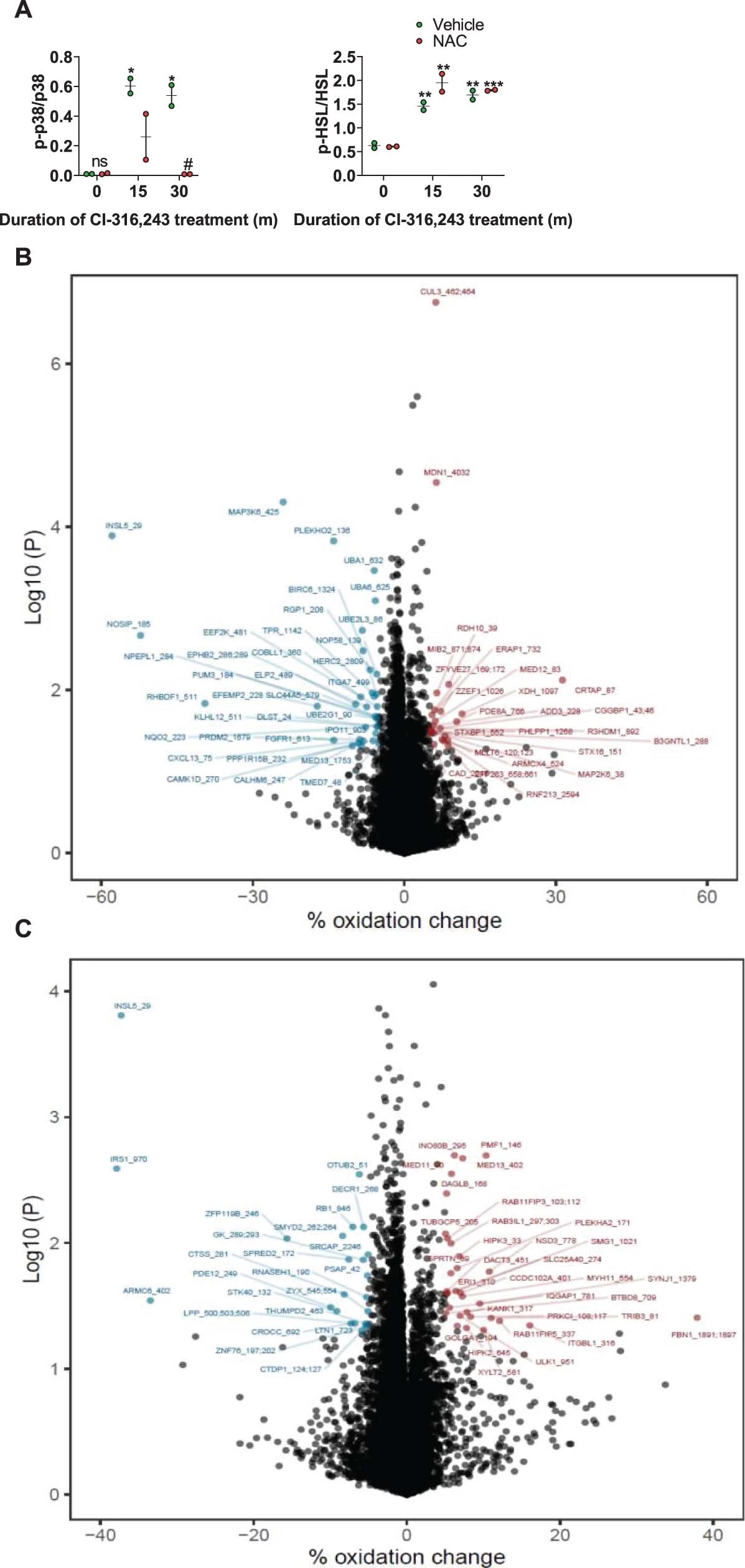
ЭМS6.ЬЧдДњаЛЯьгІ CL-316,243вдгАЯь ROS ЕФВњЩњ
5ЁЂЬЧдЪЧЪЪгІКЎРфЫљБиашЕФ
WTКЭPTG-KOаЁЪѓдкМБадРфДЬМЄЪБОљФмБЃГжКЫаФЬхЮТ(ЭМS7a)ЁЃЖјНЋаЁЪѓЛжИДЕНЪвЮТКѓЃЌЗЂЯжгыдБОЪвЮТЧщПіЯТBATжаЬЧдЫЎЦНБШНЯЃЌWTаЁЪѓBATЬЧдЫЎЦНГіЯжСЫЯджјЩ§ИпЖјPTG-KOаЁЪѓдђУЛга(ЭМS7b)ЁЃЫцКѓНјааЕкЖўДЮРфДЬМЄЃЌЗЂЯжWTаЁЪѓШдБЃГжСЫКЫаФЬхЮТЃЌЖјPTG-KOаЁЪѓдђВЛФмБЃГж(ЭМS7c)ЃЌетБэУїЬЧдДњаЛЪЧГЄЦкКЎРфЪЪгІЫљБиашЕФЁЃдкНгЯТРДЕФЪЕбщжазїепЙЙНЈСЫUcp1-creвРРЕЕФзиЩЋКЭУзЩЋжЌЗОЯИАћPTG-KOаЁЪѓ(PTG-BKO)ЃЌВЂЭЈЙ§18Ёц 7ЬьЃЌШЛКѓ4Ёц 14ЬьЕФГЄЦкРфДЬМЄгеЕМЃЈаЁБрзЂЃКРфДЬМЄЪЕбщЪБМфжмЦкЮЊ22Ёц3ЬьЃЌ18Ёц7ЬьЃЌШЛКѓ4Ёц14ЬьЃЌВЂЗЧЧАЮФЪЕбщжаМБадРфДЬМЄЕФ4ЁцгыЪвЮТRTЕФНЛЬцЃЌвђДЫгАЯьаЁгкМБадЪЕбщЁЃЃЉWTКЭPTG-BKOаЁЪѓЬЧдДњаЛЯрЙиЛљвђЕФБэДя(ЭМS7d, e)ЁЃНсЙћЯдЪОЃЌгыЖдеезщЯрБШЃЌРфДЬМЄКѓЕФPTG-BKOаЁЪѓЕФiWATжаUcp1 mRNAБэДяЯТЕї(ЭМS7f)ЁЃЧвUCP1ЕААзЫЎЦНвВЯТНЕ(ЭМS7g, S8a)ЁЃ
PTGЧУç€ВЛгАЯьUCP1дкBATжаЕФБэДя(ЭМS8b)ЁЃдк18ЁцЕЭЮТЪЪгІЪБЃЌСНзщМфЕФКФбѕСПВЂЮоВювьЃЌШЛЖјЃЌдк4ЁуCБЉТЖ3ЬьКѓPTG-KOаЁЪѓЕФКФбѕСПКЭCO2ВњСПНЕЕЭЃЌМЬајБЉТЖ4ЬьКѓЛсНјвЛВННЕЕЭ(ЭМS7h,S8c)ЁЃЮЊДЫЃЌзїепЬсГівЛИіФЃаЭЃЌМДЭЈЙ§жЌЗОЯИАћЬЧдКЯГЩКЭзЊЛЛВњЩњROSРДМЄЛюp38ЃЌЫцКѓгеЕМUcp1КЭЦфЫћВњШШЛљвђРДЕїНкФмСПЯћКФ(ЭМS7i)ЁЃ
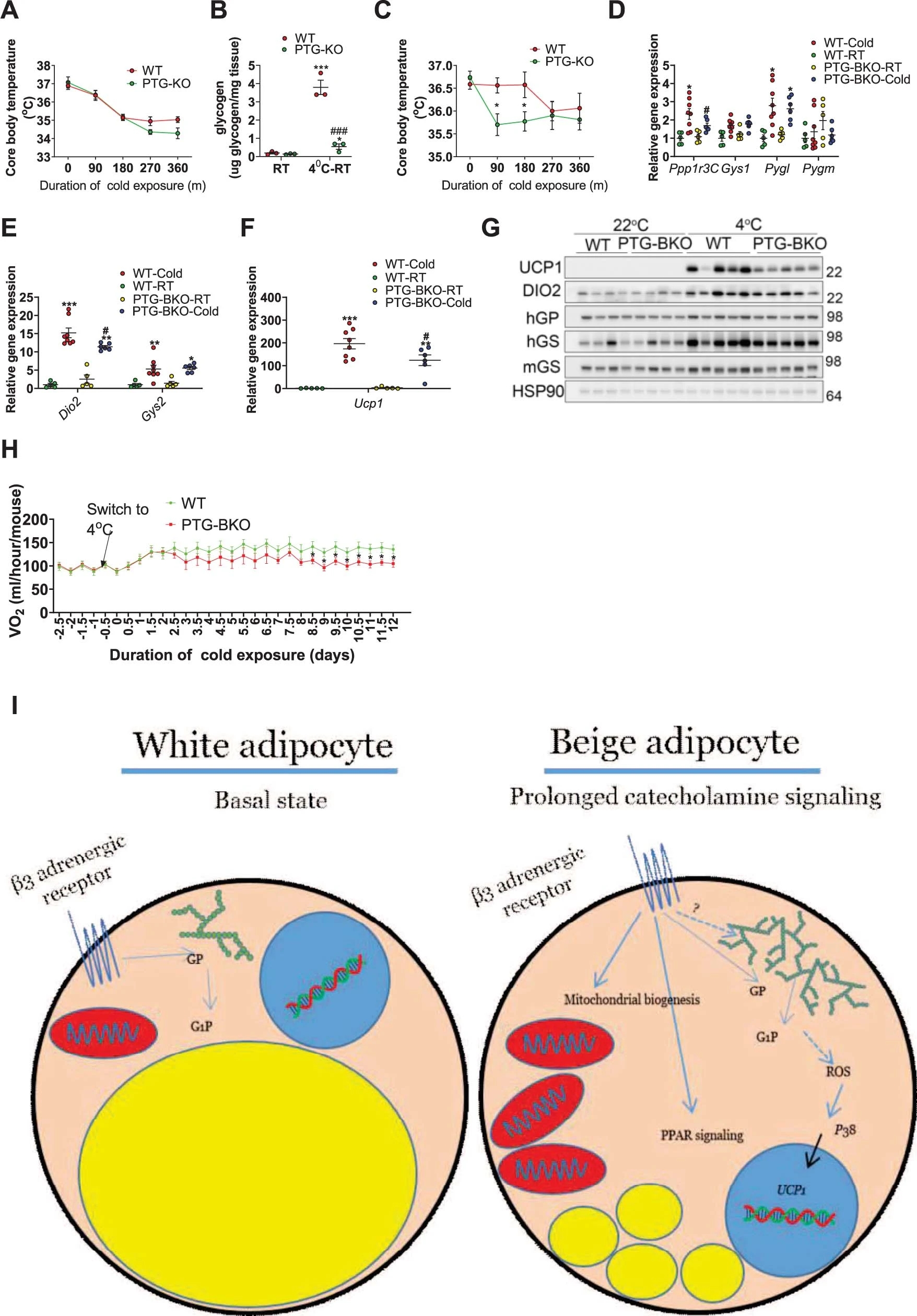
ЭМS7.PTG-KO НЕЕЭСЫГЄЦкРфДЬМЄЦкМфЕФФмСПЯћКФ
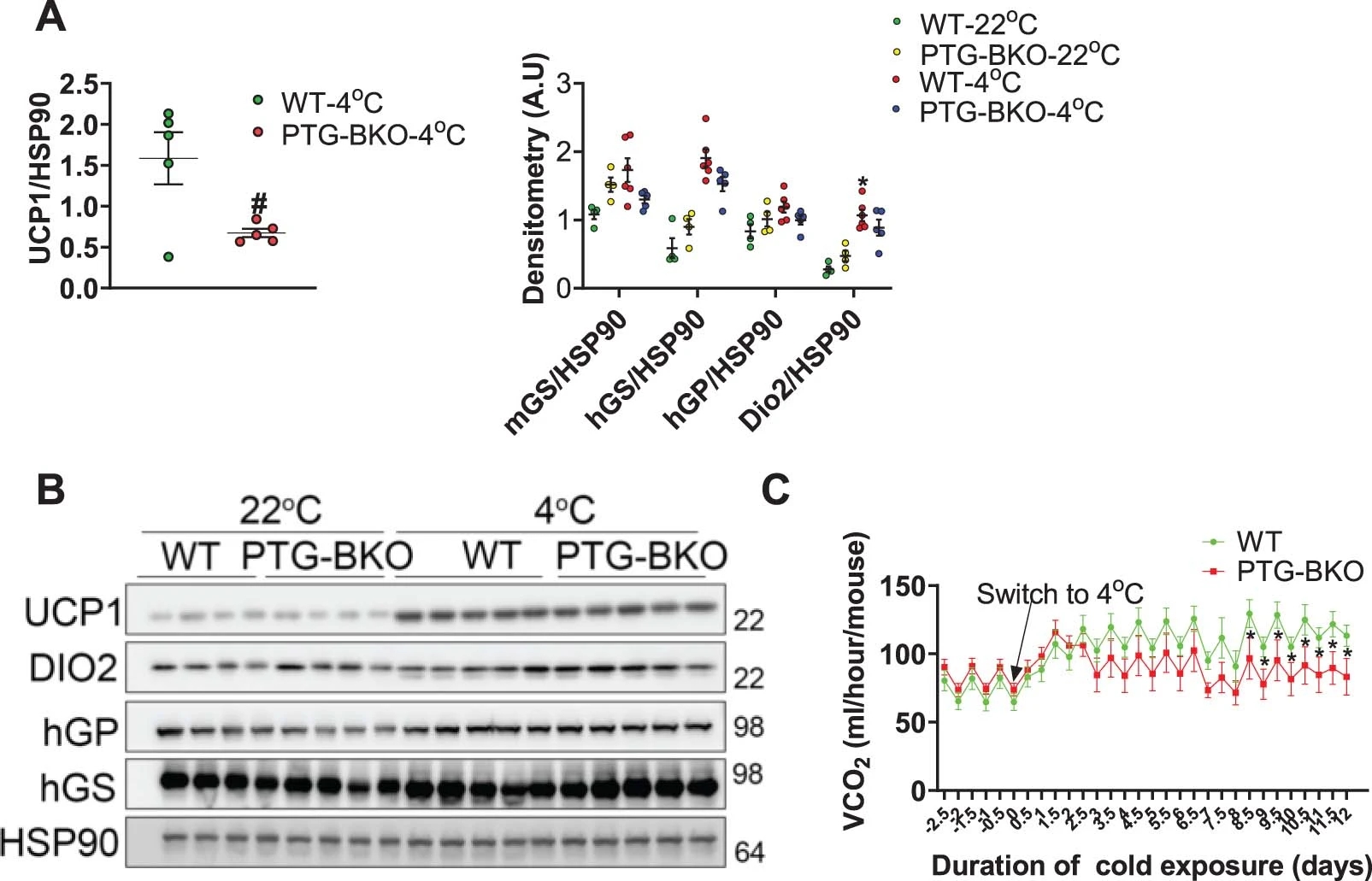
ЭМS8.ГЄЦкРфБЉТЖКѓ BAT-Ucp1 ЕФБэДядіМгВЛашвЊ PTG
змНс
ЗЪХжвбШЛГЩЮЊвЛжжШевцСїааЕФДњаЛадМВВЁЃЌЦфдвђЪЧФмСПЩуШыДѓгкФмСПЯћКФЁЃзїЮЊФмСПЕїНкЕФЙиМќЃЌЪЪгІадВњШШзїгУдкЦфжаЦ№ЕНживЊзїгУЁЃИУбаОПЗЂЯжЃЌжЌЗОзщжЏжаЕФЬЧдДњаЛЖдгкгІМЄзДЬЌЯТUCP1ЕФБэДяКЭВњШШжСЙиживЊЃЌТ§адІТ-ЩіЩЯЯйЫиаХКХЕФМЄЛюЛсгеЕМWATзиЩЋЛЏвдМАУзЩЋжЌЗОзщжЏжаЬЧдЕФЛ§РлЁЃЖјЬЧдЮШЬЌЕФЕїПиЛЙФмЙЛДйНјp38 MAPKЗЂЩњROSвРРЕадМЄЛюЃЌНјЖјгеЕМUcp1МАЦфЫћВњШШЯрЙиЛљвђЕФБэДяЃЌзюжеЕїПиКЎРфЕШгІМЄзДЬЌЯТаЁЪѓЕФФмСПЯћКФЃЌВЂМѕЧсЬхжиЁЃетвЛЗЂЯжЬсЪОСЫЬЧдДњаЛЕФЕїНкПЩФмГЩЮЊШМжЌМѕЗЪЕФаТЗНЯђЁЃ
ЙизЂЮЂаХЙЋжкКХДњаЛбЇШЫ
СЫНтИќЖрДњаЛЧАбизЪбЖ

https://wap.sciencenet.cn/blog-3483272-1318987.html
ЩЯвЛЦЊЃКДњаЛбЇШЫ--ScienceзгПЏНќЦкДњаЛОЋбЁ
ЯТвЛЦЊЃКДњаЛбЇШЫ--Cell MetabolismЃКаЁКЂзгВХзібЁдёЃЌГ§ЬЧКЭНЕжЌЃЌIsthmin-1ШЋЖМвЊ
ШЋВПзїепЕФЦфЫћзюаТВЉЮФ
- • ДњаЛбЇШЫ--Nature MetabolismЃКжЊЗёжЊЗёЃЌИЮEVsПиЬЧИпЪж
- • ДњаЛбЇШЫЁЊЁЊNature MetabolismЃКЯпСЃЬхЃКЮвСбПЊСЫ
- • КУПДЕФЩњЛЏЪщЁЊЁЊГЫЗчЦЦРЫЕФЮьЬЧСзЫс
- • Cell Metabolism: ЩНгъгћРДЗчТњТЅЃЌжЌЗОВњШШЫАЮЭЗГяЃП
- • ДњаЛбЇШЫ Cell Metabolism: ДњаЛгыУтвп ГЩвВЯєКЮАмвВЯєКЮ
- • ДњаЛбЇШЫ--NCB: ЁАШШРБЙіЬЬЁБЕФУиУмЃКЛНабжЌЗОEPAC1