博文
利用 vasp虚晶近似计算 发现联苯烯中的狄拉克点偏离高对称区域
||
元素掺杂或替代是材料设计过程中常用的一种手段,我们可以通过第一性原理计算来研究掺杂前后两种不同材料的电子性质,比如能带结构,拓扑性质等等,但我们其实也想建立一个元素掺杂或替代过程中性质连续变化的图像,计算软件VASP中提供了虚晶近似 (Virtual-crystal approximation) 计算可以实现这一功能。
我们以材料联苯烯(biphenylene)为例对其进行介绍,联苯烯是石墨烯的同素异形体,其由四元环,六元环和八元环构成,又被称为4-6-8结构,是由Fan等人于2021年首次合成出来[1],其结构如图1(a)所示。我们通过在四元环中用硼和氮替代碳[如图1(b)所示]来研究体系的电子性质变化,我们发现其电子能带结构在替代前后已经发生了很大的变化。对于C6 体系,其能带结构中存在一对第二类型的狄拉克点,被证实为狄拉克半金属[2],而对于C2(BN)2体系,相应的狄拉克点消失了,但是我们发现其存在一条位于价带与导带之间的表面态[如图2所示]。因此为了研究这条孤立表面态的起源以及狄拉克点消失后的“去向”,我们利用虚晶近似(即控制硼氮替代的比例t)的方法,在两个结构之间建立起一座“桥梁”,如图1(a→b)。

图1 (a) C6结构示意图,(b) C2(BN)2结构示意图,(c)两个结构的能带图
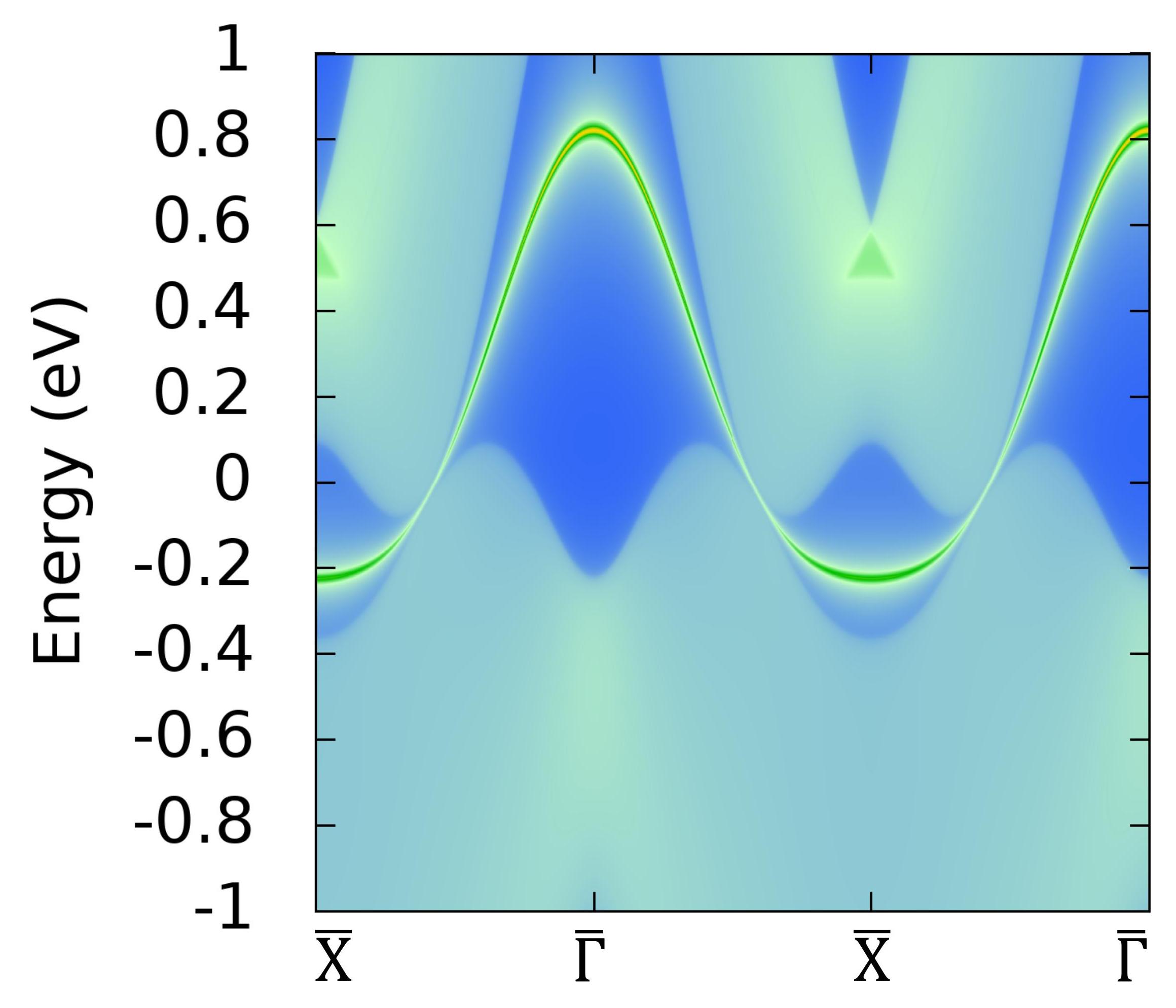
图2 C2(BN)2结构的表面态
-----------------------------------------------------------------------------------------------------------------------------------------------------------
虚晶近似计算细节
虚晶近似计算:VASP中虚晶近似是由INCAR中参数 VCA = [real array] 来控制的,在整个计算过程中只需要修改INCAR, POSCAR 和 POTCAR.
如图1(a)与(b),我们需要在两者中间设置中间结构,即我们需要在四元环中保留部分比例碳的同时,替代一定比例的硼和氮,因此对于POSCAR而言,我们需要在四元环的位置上同时放 碳和硼 以及 碳和氮 [如图3所示]。而替代的比例由INCAR中VCA参数控制,我们将四元环中碳的比例设置为20%,硼和氮的比例设置为80%。相应的POTCAR也需要根据POSCAR中的原子顺序进行修改。

图3 虚晶近似中INCAR, POSCAR 和 POTCAR相应的设置
-----------------------------------------------------------------------------------------------------------------------------------------------------------
我们通过虚晶近似计算, 如图4(a-d)所示,发现C6到C2(BN)2过程中,狄拉克点并没有直接消失,而是从高对称线Γ-Y转移到非高对称区域,与另外一对新产生的狄拉克点(在硼氮替代比例t=0.82时产生)相遇,两者湮灭在了非高对称区域。与此同时我们构建了一个紧束缚模型来描述这一个过程以及研究其表面态的演化情况,我们发现两个狄拉克点湮灭在布里渊区非高对称区域的同时,相应的表面态“黏”在一起[如图5(d→e)所示],形成了一条完整孤立的表面态,与DFT计算结果(图2)保持一致。
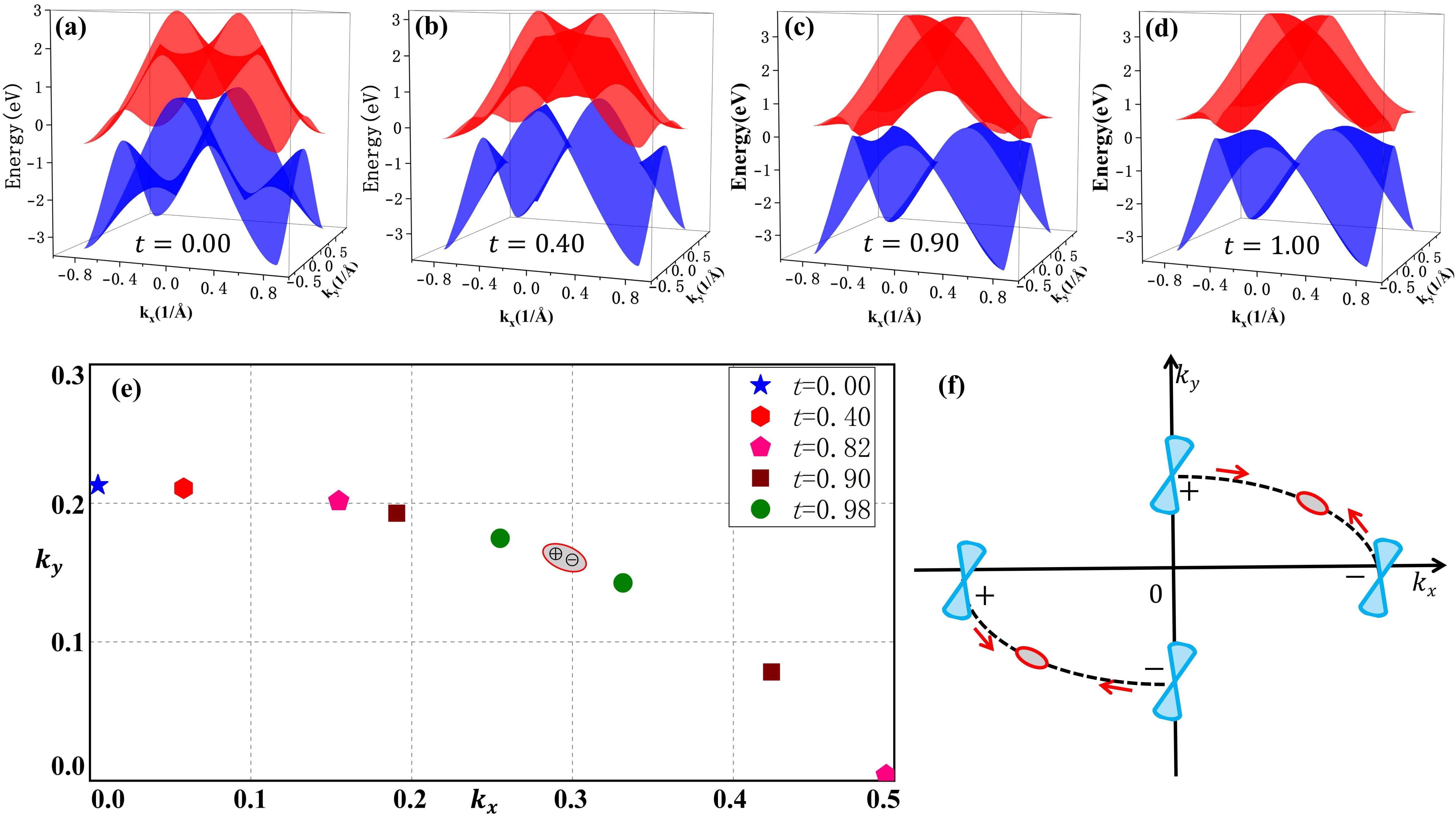
图4 (a-d)在硼氮不同替代比例下,体系导带与价带的三维能带图;(e)在不同替代比例下,能带简并点在布里渊区的位置;(f)狄拉克点移动路径的简单示意图
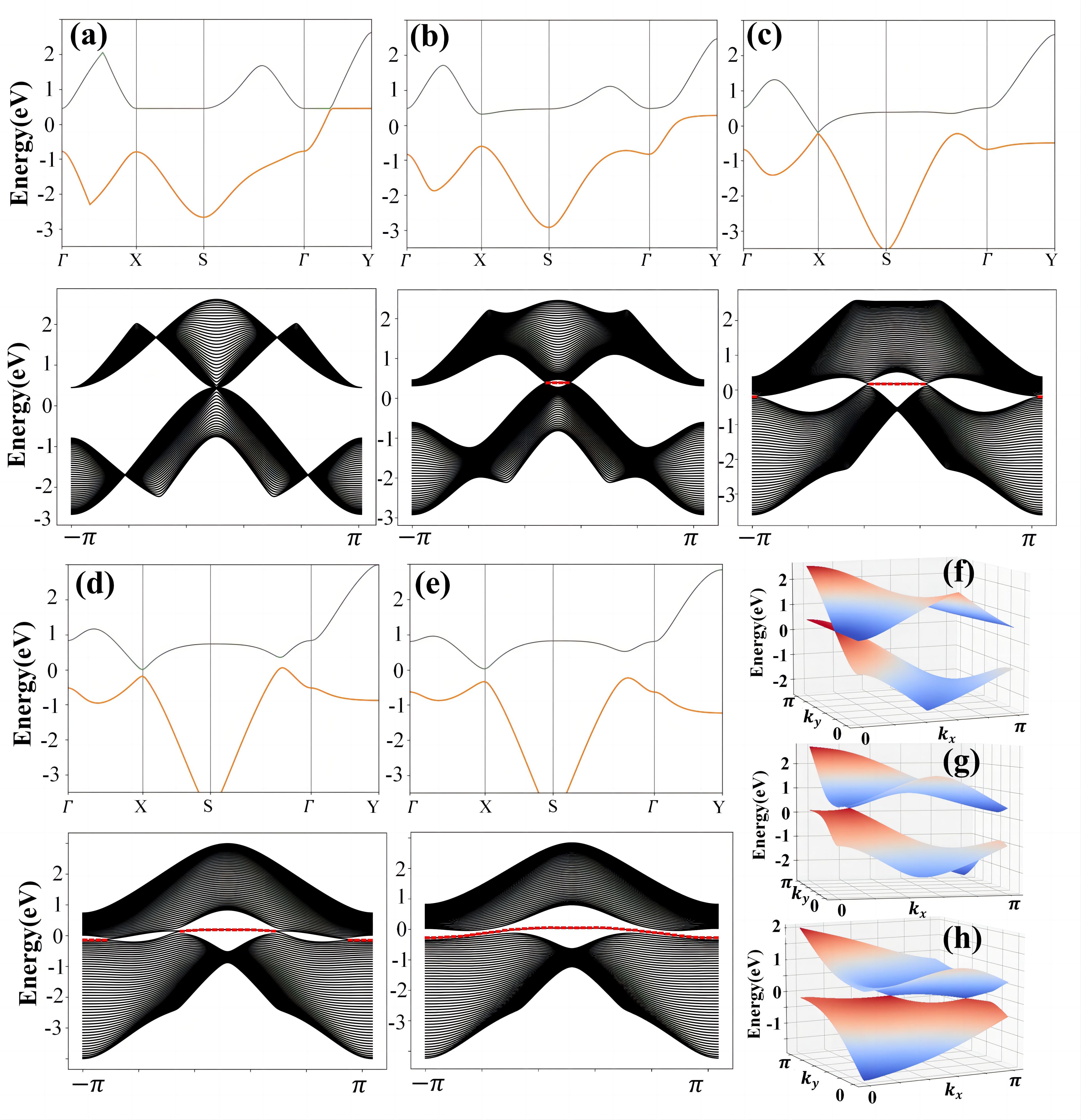
图5(a-e) 对构建的紧束缚模型进行求解能带以及表面态[3];(f-h)分别对应a,b,c情况下的三维能带图。
这一项研究[3]发现了二维材料联苯烯中的狄拉克点(能带简并点)可以偏离高对称区域移动至非高对称区域这一有趣现象。
参考文献:
[1] Q. Fan, L. Yan, M. W. Tripp, O. Krejci, S. Dimosthenous, S. R. Kachel, M. Chen, A. S. Foster, U. Koert, P. Liljeroth, and J. M. Gottfried, Biphenylene network: A nonbenzenoid carbon allotrope, Science 372, 852 (2021).
[2] P. F. Liu, J. Li, C. Zhang, X. H. Tu, J. Zhang, P. Zhang, B. T. Wang, and D. J. Singh, Type-II Dirac cones and electron-phonon interaction in monolayer biphenylene from first-principles calculations, Phys. Rev. B 104, 235422 (2021).
[3] Guang F. Yang, Hong X. Song, Dan Wang, Hao Wang and Hua Y. Geng. Topological and superconducting properties of two-dimensional C6-2x(BN)x biphenylene network: First-principles investigation[J]. Physical Review B, 2024, 109(12): 125424.
https://wap.sciencenet.cn/blog-3469498-1426844.html
上一篇:电子化合物:一类新的实空间拓扑体?
下一篇:[转载]如何根据晶体场理论分析能带轨道成分?