AlphaFold2 以令人震惊的优势取得本次蛋白质结构预测比赛(CASP)冠军,变革性地解决蛋白质折叠问题,成为本周最大的科技新闻,迅速”出圈“,一时间成为科学与工业界纷纷热议的话题。
因为笔者的专业背景,笔者能够采访到几位师友、同窗,从其各自专业角度解读和评价 AlphaFold2。袁帅博士
袁帅博士毕业自中科院生物物理所,师从饶子和院士,其代表工作是解析了一种包含多达3000个蛋白质分子的疱疹病毒衣壳(herpesvirus capsid)的结构(Yuan et al., Science 2018)。目前,他在耶鲁大学的分子生物物理与生物化学系从事蛋白质结构研究。
袁帅博士等所解析的结构。
Yuan et al., Science 2018.
12月3日,笔者就 AlphaFold2 对结构生物学的影响采访了袁帅。首先,AlphaFold2 短期内对结构生物学影响不大。2014年对结构生物学而言是一个分水岭,因为2014~15的冷冻电镜(Cryo-EM)在图像分辨率方面的突破,使得其在一些结构中获取的图像有了接近原子级别的分辨率,包括病毒、核酸、离子通道、酶复合物等,从而在小至约 150 kD 的一些结构获得分辨率约4.5 Å的结构。最近几年的进一步发展使得冷冻电镜可以解析小至 60 kD的结构。冷冻电镜的成功应用,更新了结构生物学的主要技术手段,从而很大程度上改变了结构生物学的研究对象,—— 由利用 X射线、核磁共振(NMR)等解析蛋白质晶体,转为解析单颗粒复合物。因此,如果 AlphaFold2 在2014年之前出现,一定会对整个结构生物学造成巨大冲击。第二,由于冷冻电镜等新技术的出现,结构生物学目前的工作重心是阐明功能。袁帅近期解析了新冠病毒的 Nsp1 蛋白的结构。他们的工作阐示了新冠病毒借助 Nsp1 蛋白阻碍病毒的宿主细胞合成蛋白质(Yuan et al., Mol. Cell 2020)。他们发现,Nsp1 蛋白的C端的两个螺旋结构,对阻断 mRNA 参与翻译起到重要作用。然而,另一研究组所解析的 Nsp1 蛋白结构在C端则无固定结构(unstructured)。这表明,Nsp1 蛋白的自由状态、工作状态的构象很可能不同。那么,Nsp1 蛋白在何时因为何发生了结构转变?这点,包括 AlphaFold2 在内的各种 AI 如何去捕捉呢?第三,AlphaFold2 的确可以促进结构生物学研究。目前冷冻断层成像(Cryo-ET)技术方兴未艾,但是其所获得结构的分辨率不高——结构学家们只能由此获得蛋白结构的外形轮廓。AlphaFold2 等 AI 方法显然可以此为基础,为结构学提供高精度的计算预测,即结构细节。王博申
王博申正就读于伊利诺伊大学芝加哥分校(UIC),师从梁杰博士,从事蛋白质计算化学与生物信息学研究。12月3日晚,笔者就 AlphaFold2 的技术特点采访了王博申。第一,相较于其2年前的第一个版本和其它深度学习方法,AlphaFold2 最具创新力的应该是”两张表“,即序列-残基“表”(sequence-residue edges)和残基-残基“表”(residue-residue edges)。这是在提取研究对象的特征时(feature engineering),特别着重了蛋白质折叠的物理内涵,以至于能够完成端到端(end-to-end)折叠,直接生成最终结构。这里的端到端折叠和两组edges是最重要的新概念。新的 AI 模型,即便有,相对而言,重要性也较低。第二,AlphaFold2 如何处理蛋白质结构库中的冗余结构和蛋白质序列库中的同源序列,不知道。Deepmind 自己披露的信息和演讲中展示的内容显示,他们用了蛋白质结构库(Protein Data Bank, PDB)中 170000 个结构。然而,PDB中有许多蛋白有多个结构(同一序列,不同组解出的相近结构)。现在不清楚 AlphaFold2 团队如何处理这部分冗余。通常人们会尽量在训练集内包含差异较大、同源性较低的结构,并去重,以免造成训练集内的元素权重不均匀,从而过拟合(overfit)。例如,AlphaFold2 初代版本的训练就明确提到去除了冗余。但是,本次 AlphaFold2 团队却只字不提冗余处理。另一方面,哺乳动物(如人类等)的蛋白序列的同源性较高,结构相似度也较高,与之相比,细菌等的蛋白质的同源性就低得多,同时数据也少的多。如何在训练集中选择性地囊括或者排除特定来源的蛋白,显然会对模型训练和结果造成显著影响。AlphaFold2 自己的例子(ORF8)表明,对于特定的蛋白,可能借助了近几年同源建模领域新型挖掘共进化(co-evolution)特征,而计算共进化特征通常需要较大的同源序列深度(sequence depth)。为了取得高序列深度,已有的方法一般将整个蛋白切成多个片段(比如 pfam domain),才可以得到高深度;但这样又无法涵盖一些内禀非结构片段(intrinsic disorder segment)。华盛顿大学 David Baker 组的 GREMLIN、哈佛大学 Debora Marks 组的 EVmutation等,属于这类方法。总的来说,AlphaFold2 团队目前给出的具体技术细节还太少。第三,训练 AlphaFold2 所需要的计算资源和财力是现在任何大学的科研组都无法承受的。AlphaFold2 团队提到他们使用了128 TPUv3 cores和几周的时间,并称这个计算量相对于训练一些最先进的 AI 模型并不算多(modest)。如密歇根大学安娜堡分校的张阳博士所言,这很不客观。博申用上面提到的冗余问题举了例子。为了确定在训练集中囊括或排除的蛋白,就至少需要若干次测试。为了确定 AI 模型,需要对备选的若干个模型的每一个进行若干次调参训练。若干 x 若干 x 若干,就不少了。张阳博士说可能需要200 ~ 2000次训练,博申也认为,为了获得稳定输出最佳结果的模型,训练次数应当至少以百计。我们可以估算一下费用。Google 的云端计算 TPUv3 资源,价格是 $32 / hr,“几周”按30天算:128 x 24 小时 x 30 天 x 32 = 2949120。即,训练一次模型所需计算费用约300 万美元。【如果我们计算错误,欢迎指正。】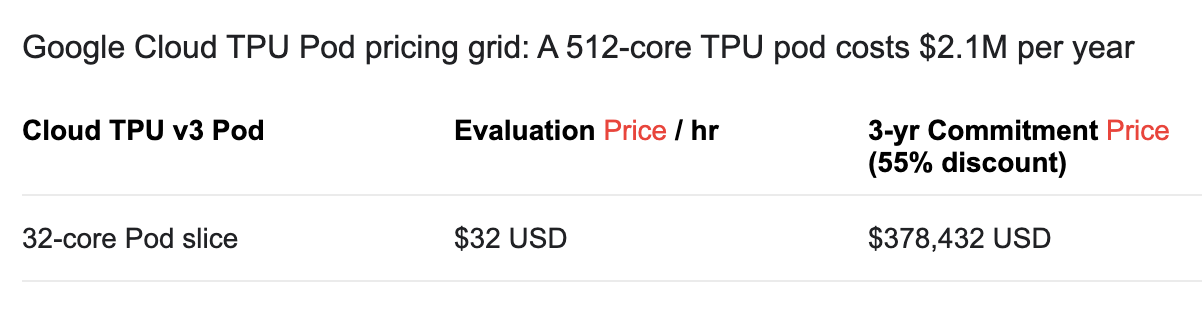
Google 的云端计算 TPUv3 资源定价。
当然,作为Google下辖部门的 Deepmind,使用谷歌的云端资源应该会便宜得多;而外部科研机构,纵然长期使用有折扣价(一年期是37% discount),每次训练也需要约200万美元。那么100次呢?
此外,还有人工费。30位作者,每人仅按20万美元计算年薪,该团队也需要600万美元经费作为工资,而这显然是低估。对美国的学术机构而言,科研组每招一个学生或博后,需要按比例与学校分成,学校分去的科研经费可高达五成。从经济角度,高校科研组与 AlphaFold2 团队完全无法争锋。第四,AlphaFold2 现下对药物设计的促进有限,有待发展。结构是理解功能,阐示功能不可或缺的部分,但仅有静态的结构(static structure),不足以弄清动态的功能(dynamic function)。对药物设计来说,药物小分子与蛋白质特定位点的结合,可能引起蛋白质构象改变,而这种改变可能还是非局域的,例如别构效应(allosteric effect)。如何在分子动力学中结合 AI 预测可能是将来 Deepmind 研究的方向。王晟博士
王晟博士毕业于中科院理论物理所,在芝加哥大学丰田研究中心(TTIC)和人类基因学系做过7年博士后研究员,期间专注于蛋白质计算化学研究,与许锦波博士合作,在将AI应用至蛋白质折叠方面做出了许多先锋性的成果,如 Ultra-Deep Learning model(Wang et al. PLOS Comput. Biol. 2017),RaptorX‐Contact(Wang et al., Proteins 2016,CASP12 冠军)。目前,王晟是腾讯 tFold 项目负责人。12月5日,笔者就 AlphaFold2 的技术特征、影响等采访了王晟。第一,AlphaFold2 技术创新在于崭新的 AI 算法,即从 1D 到 3D,直接训练出最终的结构,而非对物理特征的理解。他本人在2017年的工作(PLOS Comput. Biol. 2017),是使用 AI 深度学习折叠蛋白质的开创性工作,第一次用深度学习由 1D 训练出非常准确的2D结构。这一次,AlphaFold2 团队所使用的非常先进的 AI 算法则是划时代的,而且极具迁移性,可以潜在应用到许多领域。关于 AlphaFold2 的远超同侪的表现,王晟作了一个精妙的比喻:AlphaFold2 让他想起了《三体》中丁仪观察水滴的表面。人家 gdt-ts(全局距离检验)做到0.8,他们 0.9;人家 gdt-ts 做到 0.6,他们 0.9;人家 gdt-ts 做到 0.4,他们 0.9;人家 gdt-ts 做到 0.2,他们依然是 0.9。我以为仪器出了故障,但意识到真相后,只能大喊,“傻孩子,快跑啊!”第二,本次 AlphaFold2 的表现犹如首颗原子弹爆炸,是颠覆性、划时代性的。从业内人士的角度,这绝不是“意料之中”。正如CASP比赛创始人之一,John Moult 教授所说,“某种程度上蛋白质折叠问题已经解决了。” —— 困扰科学界50年的问题,“被一朝解决”,这本身就寓含着 AlphaFold2 的重要性。想想看,蛋白质序列库为什么比蛋白质结构库大两个数量级?因为目前还没有一个高通量解析蛋白质结构的方法,即使某个序列所对应的结构能够被结构学家解析出来,如果只信赖实验结果,那么还是必须由人工解析。从某种意义上说,在蛋白质科学领域,计算结果第一次拥有了与实验结果同等重要的地位,再也不能被轻视。AlphaFold2 对科学界和工业界的影响将极为深远。
作为AlphaFold2团队领头人 Jumper 的师弟(他是卡尔的倒数第二位博士,笔者是最后一个),笔者为这次 AlphaFold2 的表现,先失语,再震惊,而后无力,终于欢欣。
理论计算和实验从来是科学发展的两条主线,螺旋上升,交替领航。这次,AlphaFold2 令蛋白质领域的计算50年来首次追上了实验。
以上。
2020.12.5 于深圳
本文首先刊载于公众号:小王随笔 @xiaowang_essay
https://wap.sciencenet.cn/blog-3458695-1261414.html