博文
TOBIAS:基于ATAC-Seq数据的足迹分析工具
|||
众所周知,MNase-Seq、DNase-Seq、FAIRE-Seq和ATAC-Seq都是测量染色质开放性的技术。它们彼此的共同点是都能够识别染色质的某些开放区域。注意:是“某些”,而不是全部。而彼此之间的不同点以及优势领域也是很明显的。可以参考图11。
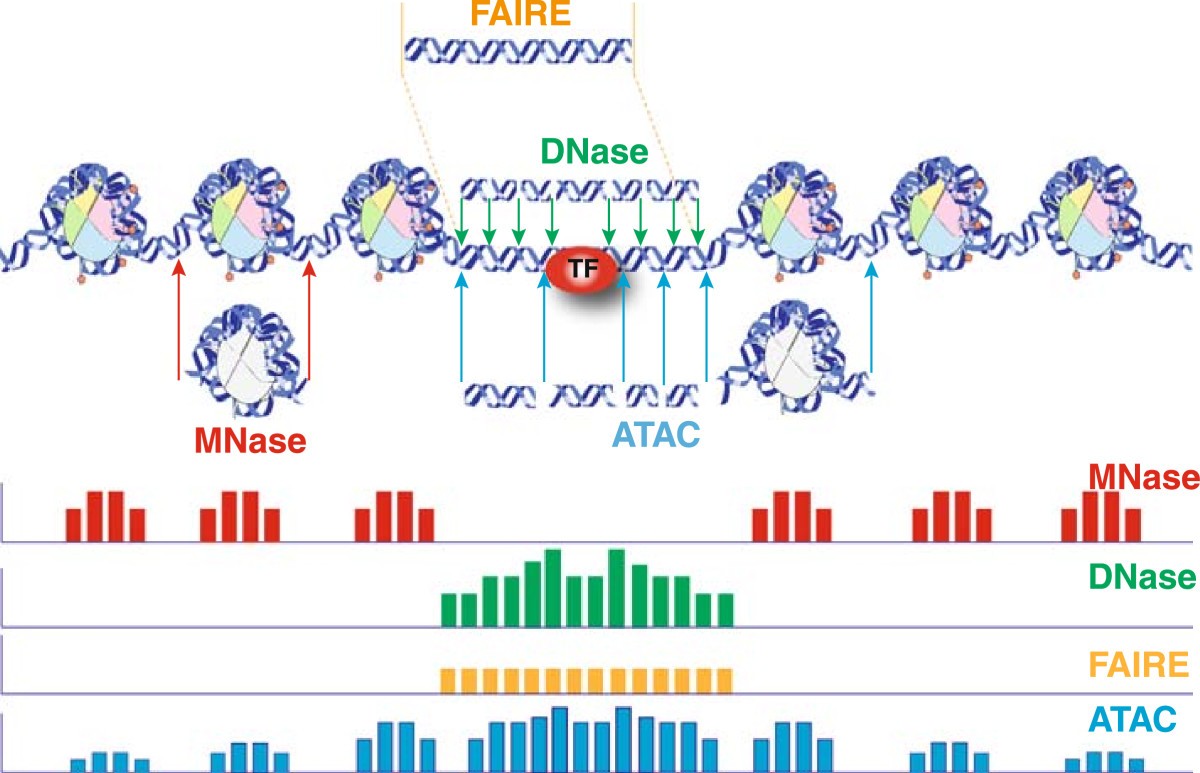
图1:MNase-Seq、DNase-Seq、FAIRE-Seq和ATAC-Seq的技术原理。
从图中可以看出,TF与裸露染色质的结合会降低染色质区域的开放性。DNase-Seq和ATAC-Seq技术都能够识别这一信息,在它的峰中会出现一个轻微的凹槽——也就是足迹(Footprint)。但由于ATAC-Seq技术的限制,凹槽的辨识度不高,难以用来做足迹分析,也就是TF与染色质的结合研究。
目前,ATAC-Seq技术凭借其低廉的价格获得广泛推广,并已经拓展到单细胞领域。ATAC-Seq代替DNase-Seq是未来的发展趋势。今天我们选取了今年八月份发表于《自然通讯》(即:Nature Communications)的一套基于ATAC-Seq数据的足迹分析流程(Framework)——TOBIAS2。由于篇幅原因,我们今天只介绍它是如何计算出这个凹槽的位置的。至于后续分析,读者可以自行查阅论文原文。
我们先回顾下ATAC-Seq的技术原理(图2)。首先,要发挥作用的染色质区域会从缠绕的组蛋白上解开,变成裸露状态。然后,我们用Tn5转座酶去切割染色质。Tn5能够特异性地识别它在染色质上的结合位点并将其切断。当然,Tn5的特异性会导致其切割位置存在偏差,但理论上我们可以认为它在染色质上是均匀用力的。其中,裸露的位置很容易被Tn5识别;而被TF结合的位置就会受到保护,很难被切割;至于组蛋白缠绕的地方,那就受到“完美庇护”,被切割的可能性非常低。然后,切下来的片段(Fragment)会经过PCR扩增和二代测序得到大量的读段(Read)。读段指的是片段两端被测序的短序列。接下来,用基因组回帖工具(例如:Bowtie3)讲过滤后的读段比对到基因组上,然后可以通过Peak-Calling工具(例如:MACS4)绘制出峰图。图2最后一步中的蓝色区域就是足迹,它无法通过ATAC-Seq数据直接得到,需要经过一系列计算得到。这就是我们今天的主题。
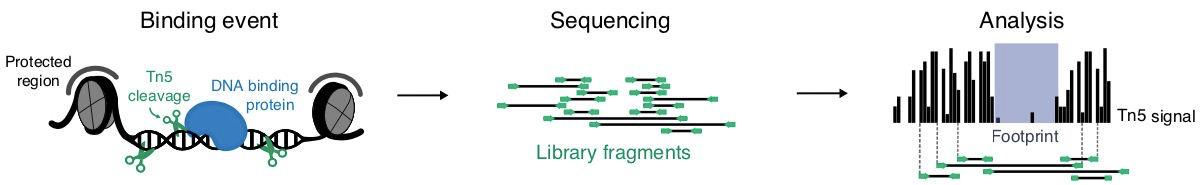
图2:ATAC-Seq技术流程。
今天我们介绍的这个TOBIAS工具需要三种输入数据:1)ATAC-Seq的测序数据,也就是读段(注意:并没有进行Peak-Calling),2)TF的模体(Motif)剖面矩阵,和 3)基因组序列注释信息(图3)。今天我们只讨论计算足迹的流程,其实只涉及到ATAC-Seq测序数据。首先,比对到基因组上的读段反映出Tn5的切割位置。但是Tn5本身具有序列特异性,也就是说即使是同样裸露的两个区域,由于序列的不同,Tn5切割的频次也不同。我们要排除这种影响,直观的方案就是估计Tn5切割位点的偏差分布,然后用实验数据生成的读段分布减去偏差进行矫正,得到矫正后的读段覆盖度,它反映了每个位置足迹存在的可能性。这里作者采用了二核苷酸权重矩阵(Dinucleotide Weight Matrix,DWM)估计出这个偏差分布5。这个偏差分布是针对基因组上的单个碱基位置的。为了将其连续化,作者估计出每个100 bp长度的窗口内部偏差的平均值。这样每个位置足迹出现的可能性已经得到了。然而,足迹是基因组上的一个个窗口,而不是单独的位置。因此,我们还有最后一步——找到这个窗口。作者采用了一个简单粗暴的方法,对窗口可能出现的位置以及窗口的大小进行穷举,最终选取足迹得分最高的那一组搭配。这里的足迹得分定义为每单位长度ATAC-Seq的每一个峰上足迹外部读段覆盖度,减去足迹内部读段的覆盖度。
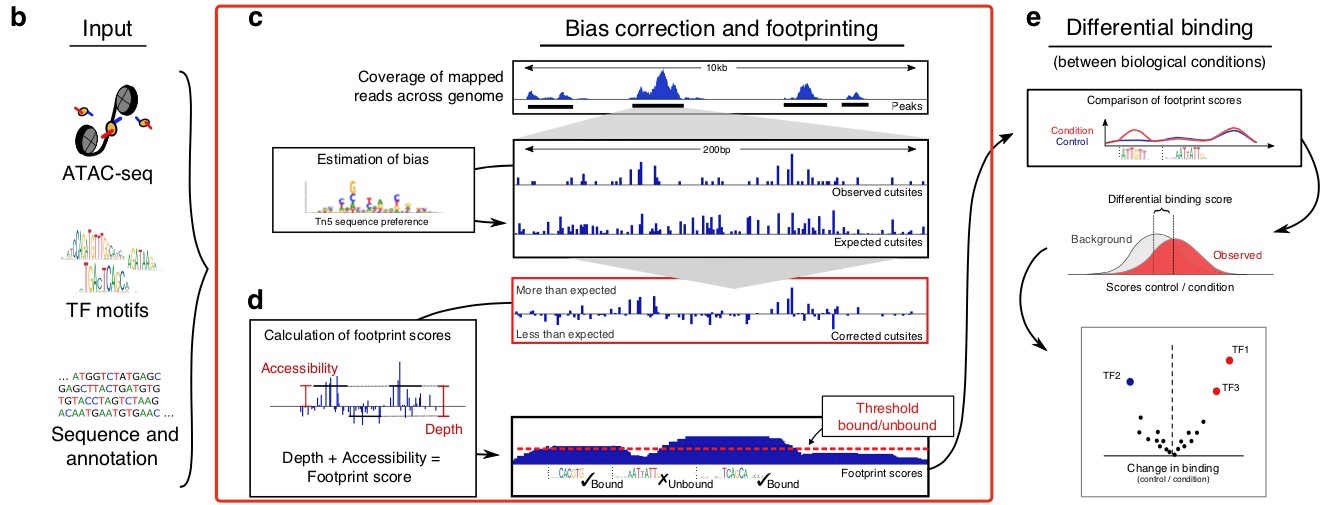
图3:TOBIAS的计算流程。
参考文献:
1. Chromatin accessibility: a window into the genome, Genome Biology (2014)
3. Scaling read aligners to hundreds of threads on general-purpose processors, Bioinformatics (2019).
4. Model-based Analysis of ChIP-Seq (MACS), Genome Biology (2008).
https://wap.sciencenet.cn/blog-3447504-1253224.html
上一篇:使用单细胞组学阐明复杂疾病中的生物网络
下一篇:关于AlphaFold的几点感受