博文
微生物多样性研究10步走(第一步)-OTU聚类与注释
|
家好,从今天起密码子将开设一个新的连载模块--微生物多样性研究10步走,内容包括在进行微生物多样性研究的过程中涉及到的知识点和常见的分析方法辨析。希望本连载能给初入研究的小白滤清思路、获得启发。
通常微生物多样性分析都是基于二代测序技术来实现的。二代测序的上机、建库等实验流程我们暂且不聊,我们从拿到下机数据来讲起。
本期先为大家分享一个基础概念--OTU。
OTU是大家进行菌群多样性测序分析的时候,不可避免需要了解的一个概念。
OTU聚类与注释
什么是OTU?为什么会出现这个概念呢?
菌群多样性分析是通过二代测序技术对微生物基因组中的marker基因(细菌为16S序列,真菌为18S或ITS序列)全长区段或部分区段进行测序从而得到环境样本中微生物种类和丰度信息的,原则上我们把测序得到的序列与数据库进行物种注释,就可以获得该序列对应的物种信息了。但是通过测序得到的序列每个样本有几万条,对每条序列都进行注释的话,工作量十分巨大,并且marker基因在扩增、测序过程中存在小部分概率测序错误,会降低后续分析准确率。所以在生信研究者们在进行多样性研究时引入了OTU的概念,来有效规避上述问题。
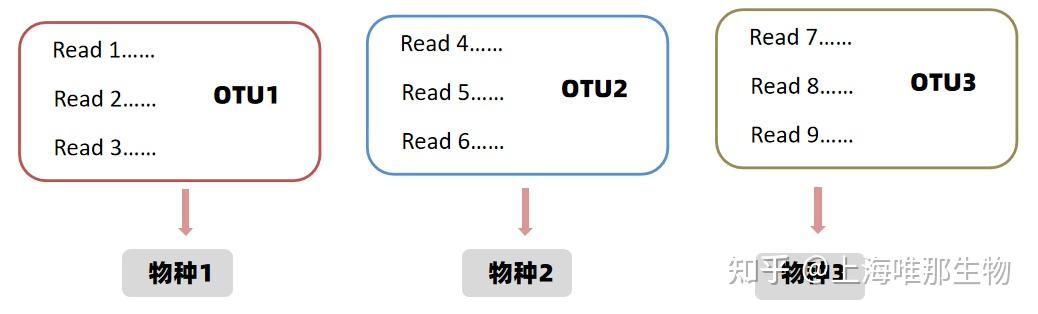
在数据下机之后,分析之前,根据序列间的相似性(通常为97%)将序列进行聚类,分成多个的分类单元,一个分类单元称为一个OTU。然后再基于OTU分类单元进行物种注释,常规方法为在OTU分类单元中选出一条代表序列,用此序列进行物种注释,得到的注释结果既为整个OTU单元的注释结果。这样不仅简化了工作量,而且OTU在聚类过程中会去除一些错误序列,提高分析的准确性与效率。
OTU聚类与注释的流程一般为:
1)提取非重复序列,降低分析中间过程冗余计算量;
2)去除单序列;
3)按照相似性进行OTU聚类,在聚类过程中去除嵌合体序列,获得OTU代表序列;
4)将OTU代表序列与数据库比对,获得OTU物种注释信息。
说到这里大家能对OTU有一个简单的认识了吧?你可以把它简单的理解为我们人为设立的一个低于种的分类学水平。(OTU的划分通常比种水平更精细)
相似性阈值如何确定?
对于OTU聚类相似度阈值很多同学也存在疑问:为什么要以97%进行相似性聚类?96%不行吗?99%不行吗?
其实在生物信息分析中我们所有涉及到的分析参数:比如聚类的相似性阈值、比对数据库的70%置信阈值等等,都是基于前期生信分析者们的开发、测试、总结得到的有较高准确性的数值。
对于相似性阈值的研究可以追溯到1973年。唐·J·布伦纳认为DNA-DNA杂交同源性大于60%的物种属于同一物种。

1994年,E. STACKEBRANDT and B. M. GOEBEL将16S rRNA相似度和DNA杂交相似度进行比较,得出16S rRNA相似度低于97%的,DNA杂交相似度都不高,所以后来科研人员们就将97%作为了默认的聚类阈值。

好的,本期分享到此结束,下期我们来聊一聊OTU抽平(同样是一个令人头秃的问题)
更多微生态相关文章:

https://wap.sciencenet.cn/blog-3445347-1283308.html
上一篇:进化分析 l cgMLST介绍(一)
下一篇:微生物菌群多样性研究—原始数据质控